NASLJEDNE BOLESTI ŽIVČANOG SUSTAVA
PREDAVANJE 16
Degenerativne bolesti s dominantnim oštećenjem neuromuskularnog aparata čine najveću skupinu svih nasljednih bolesti.
Izuzetno važni, a često i odlučujući u dijagnostici neuromuskularnih bolesti, su rezultati elektrofizioloških i biokemijska istraživanja. Jednako je velika važnost patomorfološkog nalaza. Proučavanje biopsije mišića pod svjetlosnim mikroskopom pomaže u razlikovanju miogene od neurogene atrofije. Histokemijski pregled je neophodan za otkrivanje metaboličkih lezija mišića, i elektronska mikroskopija otkrio čitavu veliku klasu bolesti – neprogresivne miopatije.
Laganim istezanjem zahvaćenih zglobova smanjuje se pojava kontraktura i samim time dovodi do sporijeg napredovanja bolesti. U tom se slučaju izvode razne specifične vježbe, a Uriasov zavoj na pritisak pokazao se učinkovitim kod poremećene dubine ekstremiteta i često se smatra kućnim tretmanom pri otpustu. Senzomotorički trening pacijenata s mišićnim bolestima provodi se u individualnoj ili grupnoj terapiji i usmjeren je na koordinaciju i finu motoriku.
Vrlo važna metoda liječenje osoba s mišićnim bolestima je ponovno vraćanje sposobnosti vožnje automobila. U tu svrhu postoji suradnja s eksternom autoškolom na odjelu za radnu terapiju, a vožnja u Bad Wildungenu odvija se u prikladnom vozilu za invalide s osposobljenim instruktorom vožnje.
Progresivne mišićne distrofije. Pojam mišićne distrofije je skupina genetski uvjetovanih poremećaja karakterizirana progresivnošću degenerativne promjene u mišićnim vlaknima bez primarne patologije perifernog (donjeg) motornog neurona.
Različiti oblici razlikuju se jedni od drugih u vrstama nasljeđivanja, vremenu početka procesa, prirodi i brzini njegovog tijeka, osobitosti topografije mišićne atrofije, prisutnosti ili odsutnosti pseudohipertrofije i povlačenja tetiva i dr. znakovi.
Dodatni pristup u rehabilitaciji mišićnih bolesti je pružanje adekvatnog savjetovanja. Ovo se ne odnosi samo na ortoze, već i na sve ostale pomagala u kući, Ciljana pomoć osobama s invaliditetom s mišićnim oboljenjima iznimno je korisna, kolica su točno izmjerena i prilagođena potrebama osoba s mišićnim poremećajima. Stolica za tuširanje, lift za kadu, invalidska kolica, rampe za invalidska kolica i drugi.
Odjel za fizikalnu terapiju koristi različite postupke za opuštanje bolesnika. Osim toga, ovaj odjel liječenja može smanjiti često manje simptome boli kod pacijenata s mišićnim poremećajima. Logoped je još jedan odjel rehabilitacije za liječenje bolesti mišića. Ovdje također treba postaviti drugu dijagnozu laringoskopijom ili određenim rendgenske studije. U kvalificiranoj rehabilitacijskoj klinici za liječenje bolesti mišića važan je preduvjet holistički terapijski pristup ovoj bolesti.
Većina mišićnih distrofija dobro je klinički proučena Detaljan opis napravljen krajem prošlog stoljeća. No, unatoč gotovo stoljetnoj povijesti proučavanja miodistrofija, pitanja njihove patogeneze i liječenja do danas ostaju neriješena. Velike se nade polažu u molekularnu genetiku, uz pomoć koje je već utvrđeno mjesto gena mnogih nosoloških oblika.
To uključuje studente diplomskog studija psihologije fakulteta. Pogođene osobe pate zbog tjelesnog nedostatka i povezanih posljedica u privatnom životu - Očekivano trajanje života često se povezuje s depresivna raspoloženja i psihosomatskih tegoba. Podrška individualnim razgovorima ili učenje o tehnikama opuštanja kao što su autogeni trening ili progresivno opuštanje mišića ovdje su od velike pomoći.
Pitanja u vezi zakona o njegovateljstvo, teški invaliditet, radno mjesto, kontakt s poslodavcem radi profesionalne rehabilitacije ili inkorporacije u tvrtku ovdje treba spomenuti kao česte teme socijalnog savjetovanja. Osim toga, često je ciljano savjetovanje u konzultacijama često postavljena pitanja za pacijente s mišićnim poremećajima. Konačno, plan terapije tijekom rehabilitacije u Bad Wildungenu također uključuje koordinaciju kvalificiranog specijaliste ortopeda. Vicerek je glavni liječnik ortopedskog odjela klinike u Hombergu u Bad Wildungenu.
Dijagnostika mišićnih distrofija često predstavlja velike poteškoće. Postoji velika varijabilnost kliničkih manifestacija, a mali broj članova obitelji otežava određivanje tipa nasljeđa.
Karakteristična motorička mana u bolesnika s mišićnom distrofijom je "patkasti" hod: bolesnik hoda gegajući se na jednu stranu. Povezan je uglavnom sa slabošću glutealnih mišića, prvenstveno srednjih i malih, koji fiksiraju zdjelicu u odnosu na femur. Kao rezultat, bolest uzrokuje nagib zdjelice prema nozi koja ne nosi (Trendelenburgov fenomen) i kompenzacijski nagib tijela u suprotna strana(Duchenneov fenomen). Pri hodu stalno se mijenja strana nagiba. Te se promjene mogu provjeriti i Trendelenburgovim testom tako da se od pacijenta traži da podigne jednu nogu, savijajući je pod pravim kutom u zglobu koljena i kuka: zdjelica na strani podignute noge se spušta (a ne diže se kao normalno) zbog slabosti gluteus medius mišića potporne noge.
Klinika provodi redovite edukacije, kao i sudjelovanje na relevantnim kongresima i simpozijima. Ovaj sastanak se posebno dogodio. Često postoji i povećanje mišićnih enzima. Bolesti se obično razvijaju polako puzajući, ali se javljaju brze bolesti. Ako sumnjate da imate miopatiju ili ste uključeni u poznatu neuromuskularnu bolest, možete zakazati termine.
Liječenje ovisi o konkretan razlog, upalne bolesti mišića mogu, na primjer, Na primjer, različiti imunoterapijski agensi. Detaljan pregled različiti genetski mišićni poremećaji mogu se pronaći u tablicama gena za neuromuskularne bolesti.
Ustajući iz vodoravnog položaja, pacijent s izraženom mišićnom slabošću proksimalnih mišića teško se prevrće na trbuh, zatim se, oslanjajući se rukama na pod, diže na sve četiri i zatim, oslanjajući se rukama na potkoljenice, zatim na kukove , postupno se uspravlja. Ovaj fenomen "biranja po svome" naziva se Gowersov manevar. Često je povezan sa slabošću mišića gluteusa maximusa.
Lijekovi koji mogu uzrokovati ili pojačati bolest mišića
Sve potrebne dijagnostičke mogućnosti dostupne su u našoj ordinaciji. U to je uključena praksa, organizirana je redovita izobrazba iz područja neuromuskularnih bolesti. Oboljeli se mogu prijaviti i u registar pacijenata. Registri pacijenata važan su prvi korak prema boljem razumijevanju bolesti i istraživanja razvoja terapije.
Oštećenje srčanog mišića
Ovdje se razmatra moguća uključenost srčanog mišića u miopatiju i druge neuromuskularne bolesti.Duchenneova miodistrofija. Pseudohipertrofična Duchenneova mišićna distrofija javlja se češće od svih ostalih bolesti mišićnog sustava (30 na 100 000 živorođene djece). Karakterizira ga rani početak i maligni tijek. Klasična slika očituje se promjenom hoda kod djeteta u dobi od 2-5 godina, u dobi od 8-10 godina djeca već teško hodaju, u dobi od 14-15 godina obično su potpuno nepokretna. Djeca imaju više ranoj dobi početni simptomi očituju se zaostatkom u motoričkom razvoju: kasnije počinju hodati, ne mogu trčati i skakati. Bolesnici umiru u 2. ili 3. desetljeću života.
Mislimo na sve neuromuskularne bolesti. Prema Waltonovoj klasifikaciji postoji 800 oblika. Ovaj simptom može biti ograničen na nekoliko mišićnih skupina ili, ovisno o bolesti, može se naći cijela muskulatura. Propadanje mišića, smanjenje mišićna masa I slabost mišića glavni su simptomi neuromuskularnih bolesti. Lijekovi se nazivaju lijekovima koji smanjuju mišićno-koštanu atrofiju mišića. Međutim, oni još ne daju izravnu dijagnozu. Niz je razloga koji mogu dovesti do djelomičnog smanjenja mišićne mase, što se dijelom odnosi na cijelo tijelo.
Jedan od prvih znakova bolesti je zbijanje mišića potkoljenice i postupno povećanje njihovog volumena zbog pseudohipertrofije. Atrofija mišića bedara, zdjeličnog pojasa često je maskirana dobro razvijenim potkožnim masnim tkivom. Proces postupno ide prema gore i širi se izvan ramenog obruča, leđnih mišića, a zatim na proksimalne dijelove ruku.
Pravi uzrok takve mišićne atrofije ili slabosti mišića može ležati u udaljenim, potpuno različitim dijelovima tijela. Uzroci mogu biti u živčanim stanicama leđna moždina, odgovoran za kretanje, u opskrbnim živcima, kada se živac prenese na mišić, ili u samim mišićima.
Liječenje neuromuskularnih bolesti
Postoje obećavajući lijekovi za liječenje miozitisa, miastenije gravis i endokrine miopatije. Prvi pristupi usporavanju mogući su s lateralna skleroza amiotrofični. U slučaju nasljednih mišićnih distrofija i spinalnih mišićnih atrofija, uzročno liječenje još nije utvrđeno. Posebna su očekivanja za budućnost usmjerena na genska terapija. Kod svih neuromuskularnih bolesti simptomatologija je dosljedna fizioterapijska potpora u kombinaciji s ortopedskim mjerama, u nekim slučajevima s respiratornom potporom, au nekim slučajevima s mogućnošću transplantacije srca.
U terminalnoj fazi slabost mišića može se proširiti na mišiće lica, ždrijela, dišne mišiće.
U uznapredovalom stadiju bolesti postoje karakteristični simptomi, kako " pačji hod”; naglašena lumbalna lordoza, pterygoid scapulae, simptom "labavog ramenog obruča". Tipične su rane mišićne kontrakture i retrakcije tetiva, osobito Ahilove tetive. Rano ispadaju refleksi koljena, a zatim refleksi s gornjih ekstremiteta.
Liječenje također uključuje kompenzaciju fizičkih ograničenja odgovarajućim sredstvima. Njemačko društvo za mišićne bolesti uspostavilo je neovisni resursni centar. Također postoji mogućnost testiranja proizvoda kod kuće u dva standardna stana ili provesti samo nekoliko dana u stanovima bez barijera.
Mišićni poremećaji Neovisni mišićni poremećaji su relativno rijetki. S druge strane, često se događa da mišiće zahvate druge osnovne bolesti, osobito bolesti živčani sustav; zarazne bolesti kao što su tifus i tuberkuloza; Parazitarne bolesti, Trinity i svinjske gliste, kao i hormonalni poremećaji.
Pseudohipertrofija se može razviti ne samo u gastrocnemius-u, već iu glutealnim, deltoidnim, trbušnim mišićima i mišićima jezika. Vrlo često srčani mišić pati od vrste kardiomiopatije. Poremećaji ritma srčane aktivnosti, širenje granica srca, gluhoća tonova, EKG promjene. Akutno zatajenje srca je najviše zajednički uzrok smrti od Duchenneove miodistrofije. Obdukcijom se nalazi fibroza i masna infiltracija srčanog mišića.
Jednostavan gubitak mišića često se javlja kao rezultat očuvanja ili odmora, Kontrakcija mišića može biti uzrokovana adhezijama ili ozljedama zbog loše cirkulacije - Kao rezultat asocijacija na gušenje. Uzrok bolova u mišićima može biti mišićna reuma, prenaprezanje, prehlada, metabolički poremećaji ili prenaprezanje pojedinih mišićnih skupina zbog deformacije skeleta.
miom - benigni tumor mišićno tkivo. Polimiozitis: Bolest mišića sa simptomima sličnim dermatomiozitisu, ali bez njih kožni osip. Što se tiče uzroka, pretpostavlja se da se povreda odnosi na autoagresije, kao na patnju koja nastaje kao posljedica napada tijela na vlastite strukture.
Često postoji kršenje motiliteta gastrointestinalnog trakta.
Smanjena inteligencija čest je simptom. Zanimljiva je činjenica da je u nekim obiteljima oligofrenija izražena oštro, u drugima relativno umjereno. Promjena viših mentalnih funkcija obično ne napreduje i nije u korelaciji s težinom mišićnog defekta. To se ne može objasniti samo pedagoškom zapuštenošću bolesne djece, koja se rano isključuju iz dječjih grupa, ne pohađaju Dječji vrtić i škola zbog motoričkih poteškoća. CT i MRI često otkrivaju cerebralnu atrofiju, vjerojatno povezanu s poremećenim prenatalnim razvojem mozga.
Postoji više od 200 oblika bolesti mišića, neki su nazvani po svojim otkriteljima, a drugi po poremećaju. Najčešće su tri podskupine. Progresivna mišićna distrofija Spinalna mišićna atrofija Neuronska mišićna atrofija. U kliničkoj praksi ove su bolesti rijetke i apsolutno su neophodne u rukama neurologa.
Genetski defekt na kromosomu 19 je lezija mišićne stanice koji se do danas ne mogu liječiti. Razlog je vjerojatno promjena membranskog sustava mišićnih stanica. Mistična distrofija može napredovati do potpunog uništenja mišićnih stanica. Najprije su zahvaćeni mišići lica, ruku, podlaktica, potkoljenica i stopala. Bolest se može javiti kod muškaraca i žena svih dobnih skupina.
Djeca često razvijaju adiposogenitalni sindrom, ponekad i druge znakove endokrine insuficijencije. Često se nalaze promjene u koštanom sustavu: deformacija stopala, prsa, kralježnica, difuzna osteoporoza.
Posebnost Duchenneov oblik je visok stupanj hiperenzimemija već na rani stadiji razvoj procesa. Dakle, razina enzima specifičnog za mišićno tkivo - kreatinin fosfokinaze - u krvnom serumu može premašiti desetke, pa čak i stotine puta normalne performanse. Oštar (10-100 puta) porast kreatinin fosfokinaze (CPK) u neuromuskularnoj patologiji trebao bi potaknuti raspravu prvenstveno o sljedećim bolestima: Duchenneova bolest, Beckerova bolest, poliomiozitis i dermatomiozitis, paroksizmalna mioglobulinurija, distalna miodistrofija. Tek u uznapredovalim stadijima bolesti stupanj hiperenzimemije se postupno smanjuje. Postoje izvješća o povećanju CPK u fazi intrauterinog razvoja.
Odgođeno opuštanje mišića tipično je nakon napetost mišića. Posljedice su slabost mišića i ograničenje pokreta u nogama, rukama i šakama, pogoršanje fine motorike. Neki bolni mišići posebno su jaki zbog tipičnog sastava tkiva ispod njih mišićna vlakna ugrađen u masno i vezivno tkivo.
Također se naziva i zaraza leđne moždine mišićni spazam. Ima ih do 30 razne forme spinalna mišićna atrofija. Najčešći oblik je proksimalna spinalna mišićna atrofija, opisana ovdje. Označava se kako slijedi nakon početka trupa.
Duchenneova mišićna distrofija prenosi se na X-vezan recesivan način. Gen se nalazi na kratkom kraku X kromosoma. Učestalost mutacije gena je prilično visoka (30%), što objašnjava veliki broj sporadični slučajevi.
Mutacija (najčešće brisanje) dovodi do spolnog ili gotovo potpunog izostanka genskog produkta - strukturnog proteina distrofičara. Fiziološka uloga distrofija nije u potpunosti utvrđena. Nalazi se u visokim koncentracijama u sarkolemi, očito igrajući ulogu u održavanju integriteta ove membrane. Odsutnost distrofije uzrokuje strukturne promjene u sarkolemi, što pak dovodi do gubitka intracelularnih komponenti i povećanog ulaska kalcija, što u konačnici dovodi do smrti miofibrila. Vjeruje se da je nedostatak distrofičnih u sinaptičkim zonama kortikalnih neurona uzrok mentalne retardacije.
Razlog je vjerojatno genetski defekt. Nervne ćelije u leđnoj moždini inficirane su prednje skvamozne stanice. Pretpostavlja se da je uzrok živčani sustav živaca. Ovo se odnosi samo na motorički živčani sustav. Dijelovi živčanog sustava koji su odgovorni za osjet dodira, percepciju boli i temperaturu ostaju netaknuti. Funkcija Mjehur a rektum se ne pogoršava.
Oznaka također predstavlja gubitak mišića zbog živaca. Uzrok je gotovo uvijek genetski defekt, ovojnice živčanih vlakana postaju abnormalno debele ili su same ovojnice živaca uništene. Na živčana vlakna utječe na ruke i stopala.
Za medicinsko genetičko savjetovanje vrlo je važno utvrđivanje heterozigotnog nositelja. Uz Duchenneovu miodistrofiju u heterozigota, u približno 70% slučajeva, subklinička, a ponekad čak i jasni znakovi patologija mišića - određeno zbijanje, pa čak i povećanje mišića potkoljenice, brzi umor mišića tijekom tjelesnog napora, promjene u EMG-u i patomorfološki pregled uzoraka biopsije mišića. Najčešće heterozigotni nositelji pokazuju povećanje aktivnosti kreatinin fosfokinaze.
Usporava se intenzitet brzine provođenja živaca. Počinje u donjim ekstremitetima s gubitkom mišića i popratnom slabošću mišića. Simptomi zatim rastu u donjim ekstremitetima, kasnije zahvaćajući ruke i podlaktice. Senzorni poremećaji su niski. mogući su autonomni poremećaji kao što je previše ili premalo znojenje i poremećaj temeljnog krvotoka. Mogu se pojaviti manji spastični simptomi u nogama.
Slabost mišića proizlazi iz slabosti mišića sve dok mišići ne budu potpuno funkcionalni, što rezultira značajnim teškim ograničenjima pokreta u nogama, rukama i rukama. Mišići su vrlo važan dio našeg tijela. Bez mišića tijelo gubi sposobnost kretanja i obavljanja raznih radnji. Zapravo, bez ljudskog mišićni sustav vjerojatno nećete moći preživjeti. To je zbog činjenice da većina organa u probavni sustav sastoje se od mišića, pa čak i samo srce, koje pumpa krv, također je mišić.
U slučaju kliničke slike Duchenneove miodistrofije u žena, prvo treba isključiti mogućnost anomalije na X kromosomu - Shereshevsky-Turnerov sindrom (XO), Morrisov sindrom (XY) ili mozaicizam u tim sindromima.
Duchenneova mišićna distrofija, koja se počinje razvijati još u prenatalnom razdoblju, u biti je kongenitalna miopatija i može se dijagnosticirati ubrzo nakon rođenja izvođenjem biopsije mišića i određivanjem aktivnosti CPK.
Beckerova miodistrofija. Uz teški, maligni oblik X-vezane Duchenneove miodistrofije, postoji benigni oblik - Beckerova bolest. Što se tiče kliničkih simptoma, vrlo je sličan Duchenneovom obliku, ali u pravilu počinje kasnije - u dobi od 10-15 godina, teče lagano, pacijenti ostaju radno sposobni dugo vremena, u dobi od 20 godina. -30 godina i kasnije još mogu hodati. Plodnost nije smanjena, pa se bolest ponekad prati u nekoliko generacija obitelji: bolestan čovjek prenosi bolest na svog unuka preko svoje kćeri ("djed efekt"). Početni simptomi, kao i kod Duchenneove bolesti, manifestiraju se slabošću mišića zdjeličnog obruča, zatim proksimalnih donjih ekstremiteta. Bolesnici mijenjaju hod, imaju poteškoća pri penjanju uz stepenice, pri ustajanju s niskog sjedala. Karakterizira ga pseudohipertrofija mišića potkoljenice. Retrakcija kalkanealne (Ahilove) tetive manje je izražena nego kod Duchenneove bolesti.
S ovim oblikom nema intelektualnih oštećenja, kardiomiopatija je odsutna ili blago izražena.
Kao i kod drugih X-vezanih miodistrofija, Beckerov oblik značajno povećava aktivnost CPK, iako u manjoj mjeri nego kod Duchennea, ne prelazeći 5000 jedinica. Gen za Beckerovu bolest, kao i Duchenneovu bolest, lokaliziran je u kratkom kraku X kromosoma; vjerojatno je da su oba lokusa blisko povezana ili su alelni. Za razliku od Duchenneove bolesti, kod koje praktički nema distrofije, kod Beckerove bolesti se sintetizira abnormalna distrofija. Razlike se također nalaze u biopsiji mišića. Kod Beckerove mišićne distrofije mišićna vlakna obično nisu okrugla, hijalina vlakna, karakteristična za Duchenneovu mišićnu distrofiju, iznimno su rijetka.
Landouzy-Dejerine miodistrofija (miodistrofija lica i ramena). Bolest se prenosi autosomno dominantnim putem s visokom penetrantnošću, ali donekle promjenjivom ekspresijom. Javlja se znatno rjeđe od Duchenneove miodistrofije (0,4 na 100 tisuća stanovnika). Vjeruje se da je gen za ovu bolest lokaliziran na 4. kromosomu. Žene obolijevaju češće od muškaraca (3:1), Tjelesno preopterećenje, intenzivni sportovi, kao i neracionalno bavljenje fizioterapija može pridonijeti težem tijeku bolesti.
Landouzy-Dejerineova miodistrofija je relativno povoljan trenutni oblik mišićne patologije. Počinje u dobi od oko 20 godina, ponekad i kasnije. Međutim, u obiteljskim slučajevima bolesti, kada je moguće pratiti dinamiku mlađih članova obitelji, moguće je uočiti slabost mišića, primjerice mišića lica, i to u ranijoj dobi. .
Slabost i atrofija mišića najprije se javlja u mišićima lica odn pojas za rame. Postupno se ti poremećaji šire na mišiće proksimalnih ruku, a potom i na Donji udovi. U većini slučajeva najprije su zahvaćeni mišići prednje površine nogu (s razvojem visećeg stopala), zatim mišići proksimalnih nogu. Na vrhuncu bolesti teško su zahvaćeni kružni mišići oka i usta, pectoralis major, anterior serratus i donji dijelovi trapezius mišića, latissimus dorsi, biceps, triceps mišići ramena. karakteristika izgled pacijenti: tipično lice miopat s "poprečnim osmijehom" ("La Giocondin osmijeh"), protruzija Gornja usna("tapirske usne"), izražene pterigoidne lopatice, osebujna deformacija prsnog koša s njegovim spljoštenjem u anteroposteriornom smjeru i rotacijom unutar ramenih zglobova. Često postoji asimetrija lezije, čak i unutar jednog mišića (npr. orbicularis oculi). Može se uočiti pseudohipertrofija gastrocnemiusa, deltoidnih mišića, a ponekad i mišića lica. Kontrakture i retrakcije su umjereno izražene. tetivni refleksi Dugo vrijeme održavaju se, ali ponekad opadaju u ranoj fazi.
Znakovi oštećenja srčanog mišića su rijetki. Aktivnost enzima u serumu blago je povećana i može biti normalna. Intelekt ne pati. Očekivano trajanje života u većini slučajeva nije smanjeno. Zanimljiva je činjenica da EMG kod Landouzy-Dejerine miodistrofije često nije sasvim tipičan za mišićnu razinu lezije. U nekih bolesnika (članova iste obitelji) može se primijetiti smanjenje amplitude biopotencijala, interferencijske vrste krivulje, u drugih, naprotiv, smanjenje frekvencije i hipersinkrone aktivnosti, ponekad s tipičnim piketanjem. ogradni ritam. Treba imati na umu spinalnu varijantu, koja oponaša Landouzy-Dejerineovu bolest.
Erb-Rothova miodistrofija (miodistrofija udova i pojasa). Prenosi se autosomno recesivno, oba spola su jednako zahvaćena. Početak bolesti u većini slučajeva odnosi se na sredinu 2. desetljeća života (14-16 godina), no opisuje se kao rani, pseudo-Duchenneov oblik, kada se prvi simptomi javljaju prije 10. godine života i bolest je teška, kasna varijanta s početkom nakon 30 godina.
Tijek bolesti može biti brz ili sporiji, u prosjeku potpuna invalidnost nastupa unutar 15-20 godina od pojave prvih simptoma. Miodistrofija počinje ili oštećenjem mišića zdjeličnog obruča i proksimalnih nogu (Leiden-Mobiusov oblik), ili iz ramenog obruča (Erbov oblik). U nekim slučajevima istovremeno su zahvaćeni rameni i zdjelični pojas. Mišići leđa i trbuha prilično pate. Bolesnici imaju karakterističan "pačji" hod, teško se dižu iz ležećeg i sjedećeg položaja, naglašeno lumbalna lordoza. Mišići lica u većini slučajeva nisu zahvaćeni. Za ovaj oblik kontrakture i pseudohipertrofija nisu karakteristične. Mogu se pojaviti terminalne atrofije i retrakcije tetiva. Inteligencija je obično očuvana. Srčani mišić uglavnom nije zahvaćen. Razina enzima u krvnom serumu u pravilu je povećana, ali ne tako oštro kao kod X-vezane miodistrofije. Postoje indicije da je u muških bolesnika razina CPK viša nego u ženskih bolesnika. Postoji značajna razlika u ekspresiji mutiranog gena u različitim članovima obitelji - uz teške klinička slika mogu postojati relativno blagi pa čak i izbrisani klinički simptomi. Smrt obično nastupa od plućnih komplikacija.
Budući da je klinika miodistrofije udova i pojasa posebno spremna imitirati neuromuskularne bolesti različite prirode, potrebno je, osobito u sporadičnim slučajevima i kod kasni početak bolesti, provesti temeljit klinički pregled kako bi se isključila spinalna amiotrofija, polimiozitis, metaboličke, endokrine, toksične, medikamentozne, karcinomatozne miopatije. U prošlosti je postojala jasna pretjerana dijagnoza ovog oblika mišićne distrofije.
Liječenje mišićnih distrofija. Terapijske mogućnosti za mišićne distrofije vrlo su ograničene. Etiološki i patogenetsko liječenje praktički nepostojeći. Simptomatsko liječenje prvenstveno je usmjereno na sprječavanje razvoja kontraktura, održavanje postojeće mišićne snage i, eventualno, na određeno smanjenje stope atrofije. Glavni zadatak je maksimizirati razdoblje tijekom kojeg se pacijent može samostalno kretati, budući da se kontrakture, skolioza i respiratorni poremećaji brzo povećavaju u ležećem položaju. Medicinski kompleks treba uključiti terapeutske vježbe, masažu, ortopedske mjere, terapiju lijekovima.
Terapeutska gimnastika sastoji se od pasivnih i aktivnih pokreta koji se izvode u svim zglobovima u različitim položajima: stojeći, sjedeći, ležeći, s različitim položajima udova. aktivni pokreti Poželjno je izvoditi u izometrijskom načinu rada. Gimnastiku treba raditi redovito nekoliko puta dnevno. Istodobno, treba upozoriti na pretjerane vježbe, osobito one praćene prenaprezanjem mišića. Važne su (osobito nakon imobilizacije bolesnika) vježbe disanja.
Ortopedske djelatnosti konzervativne (specijalne udlage) i operativne prirode (ahilotomija, transekcija gastrocnemius mišića), usmjerene na ispravljanje kontraktura i novonastalih patoloških postavki ekstremiteta, također imaju za cilj očuvanje mogućnosti samostalnog kretanja. U svakom slučaju potrebno je pojedinačno odvagnuti očekivane koristi i moguće štete od kirurške intervencije. Treba imati na umu da je često (osobito kod teške hiperlordoze i slabosti mišića kvadricepsa femorisa) ekvinovarusni položaj stopala od kompenzacijske važnosti, a nakon, na primjer, ahilotomije, pacijent može biti potpuno imobiliziran. S razvojem kontraktura, preporuča se pažljivo istezanje mišića do 20-30 puta dnevno, nakon čega slijedi udlaga tijekom spavanja.
Terapija lijekovima uključuje imenovanje metaboličkih lijekova usmjerenih na popunjavanje nedostatka energije i proteina, ali njihova učinkovitost je vrlo upitna. Koriste se antagonisti kalcija (zbog defekta utvrđenog kod Duchenneove bolesti stanične membranešto dovodi do povećanog unosa kalcija u stanicu), imunomodulatori, spojevi koji sadrže fosfor (ATP, fosfaden), vitamin E (100 mg oralno 3 puta dnevno). Dokazano je da primjena prednizolona (0,75 mg/kg dnevno) u Duchenneovoj bolesti može dramatično povećati snagu mišića, ali taj učinak traje najviše godinu dana i općenito ne utječe na ishod bolesti. Zbog ozbiljnih nuspojave, nastajanje vri dugotrajnu upotrebu lijek, njegova je uporaba neprikladna. Procjene učinka anabolički steroidi kontroverzni i njihovo je imenovanje često povezano s neopravdanim rizikom. Pri procjeni učinka određenih lijekova u Duchenneu, treba imati na umu da s umjerenom težinom bolesti u bolesnika u dobi od 3-6 godina može doći do relativne stabilizacije stanja povezanog s razvojem vezanim uz dob. mišićnog sustava, stjecanje motoričkih vještina, koje mogu donekle privremeno nadoknaditi tekući distrofični proces.
Od određene važnosti je korekcija prehrane bolesnika, preporuča se dijeta bogata bjelančevinama i malo masti te smanjeno kalorijska s optimalnim sadržajem vitamina i mikroelemenata. Važnu ulogu ima psihološka podrška bolesniku, nastavak edukacije i pravilna profesionalna orijentacija.
Stranica 44 od 44
Uključeni su skeletni mišići patološki proces s raznim degenerativnim, metaboličkim i upalne bolesti. U većini slučajeva to rezultira degeneracijom mišićnih vlakana, au kroničnim oblicima njihovom zamjenom. vezivno tkivo i masti. Značajnije su oštećene proksimalne mišićne skupine nego distalne, kao i donji udovi u odnosu na gornje. Bolesno dijete odlikuje se takozvanim pačjim (gegajućim) hodom, ne može trčati, penjati se stepenicama i ustati ako je u sjedećem položaju. Njegovi tetivni refleksi su depresivni, stupanj njihovog izumiranja proporcionalan je stupnju slabljenja mišićne snage. Osjetljivost nije pogođena.
Dijagnostički vrijedne laboratorijske metode uključuju određivanje aktivnosti enzima, posebice kreatin-fosfokinaze, u serumu. Ovaj enzim, koji katalizira reakciju: fosfokreatin + ADP-kreatin + ATP, prisutan je uglavnom u moždanim stanicama i mišićnom tkivu. U nekim difuznim mišićnim bolestima, osobito kod mišićne distrofije, njegove prekomjerne količine prodiru u međustanični prostor i krv. U bolesnika je obično povećana aktivnost serumske laktat dehidrogenaze i glutamin oksaloctene transaminaze, ali njihova široka rasprostranjenost u drugim tkivima, uključujući jetru, smanjuje specifičnost testa. Obično je za razjašnjenje dijagnoze potrebna biopsija mišićnog tkiva.
Upalne bolesti mišića. Upala mišićnog tkiva prati neke infekcije, posebice trihinelozu, toksoplazmozu i one uzrokovane Coxsackie virusom. Često je sastavni dio kolagenskih bolesti, uključujući dermatomiozitis, eritematozni lupus, periarteritis nodosa i reumatoidni artritis.
Polimiozitis. Difuzna izolirana upala mišića nepoznate etiologije naziva se polimiozitis. Karakterizira ga brzo progresivni tijek, slabost i bol u proksimalnim mišićnim skupinama. Često su mišići vrata uključeni u proces, pa je djetetu teško podići glavu i zadržati je u tom položaju. Laboratorijski znakovi upale mišića uključuju povećanje ESR i broja leukocita. Međutim, njihov nedostatak ne isključuje polimiozitis. Razine serumskih enzima obično su povišene. U biopsiji mišića utvrđuje se degeneracija i djelomična regeneracija vlakana i njihova infiltracija limfoidnim stanicama. Teško je razlikovati polimiozitis od mišićne distrofije i dermatomiozitisa. Može predstavljati atipični oblik dermatomiozitisa, iako je histologija dvaju stanja donekle različita: dermatomiozitis je karakteriziran vaskulitisom, kojeg kod polimiozitisa obično nema. Prognoza za potonje je nešto povoljnija. Liječenje kortikosteroidima prati učinak, ali kada se ponište, može doći do recidiva.
Progresivni osificirajući miozitis. Etiologija ove rijetke bolesti vezivnog tkiva i mišića nije poznata. Zabilježeno je da od nje boluju braća i sestre, uključujući blizance, a prenosi se na krvne srodnike u ravnoj liniji. Smatra se da se bolest nasljeđuje autosomno dominantno. Dječaci obolijevaju 2-3 puta češće od djevojčica.
Patološki znakovi ovise o stadiju bolesti. U ranim fazama, lokalni edem i infiltrati upalnih stanica nalaze se u mišićima i tetivama. Kasnije se područja upale zamjenjuju granulacijskim tkivom, a na kraju se u lezijama formiraju područja hrskavice i koštanog tkiva.
Gotovo 75% bolesne djece ima urođene mane razvoj, najčešće nerazvijenost prstiju i ankiloza falangi prvih prstiju i nerazvijenost prvih prstiju, polidaktilija, zakrivljenost prstiju, sindaktilija (noge), deformacija ušne školjke gluhoća, nedostatak zuba. Iste kongenitalne malformacije mogu biti i kod rođaka bolesnika koji nemaju progresivnu bolest vezivnog tkiva i mišića. Dob u kojoj myositis ossificans može početi varira od rođenja do starijeg djetinjstva. Obično se razlikuju tri stupnja: 1) ograničena, često topla i na dodir mekana tjestasta oteklina mekih tkiva pojavljuju se na mjestima manjih lokalnih ozljeda; 2) nakon nekoliko dana simptomi upale nestaju, a lezija otvrdne; 3) dolazi do okoštavanja zahvaćenog područja. Povremeno se pojavljuju nove lezije, uglavnom na vratu i leđima. primarni simptom tortikolis može postati ako se proces razvio u sternokleidomastoidnom mišiću. Na kraju se okoštavanje proteže na mnoge tetive i ligamente. Nastaje ankiloza kralježnice i zglobova ruku i nogu (slika 21-5). Upala se može proširiti na temporomandibularne zglobove, što otežava žvakanje. Koštane izrasline mogu stršiti kroz kožu. U adolescenciji bolest često dovodi do potpune imobilizacije i smrti zbog zatajenje disanja i prestanak disanja, iako postoje izvještaji o slučajevima preživljavanja. Kod osificirajućeg miozitisa postoji visok rizik od razvoja osteogenog sarkoma.
Riža. 21-5. Dijete s progresivnim myositis ossificans (tipično držanje s ukočenošću vrata i leđa).
Ponekad je patološki proces ograničen na mjesto prethodne ozljede mekog tkiva (miositis ossificans circumscripta). Raširena kalcifikacija mišićnog tkiva također se može pojaviti kod kroničnog polimiozitisa i dermatomiozitisa.
rezultate laboratorijske metode studije nemaju dijagnostičku vrijednost.
Serumske razine kalcija, fosfora, alkalne fosfataze, kao i aktivnost kreatin fosfokinaze i drugih enzima ostaju normalni. Kost u fokusu oštećenja ne razlikuje se u strukturi od norme.
Postojeće metode tretmani nisu zadovoljavajući. U nekim slučajevima zabilježeno je usporavanje razvoja bolesti uz primjenu ACTH i drugih kortikosteroida. Upitna je njihova uloga u konačnom rezultatu liječenja.
Endokrine i metaboličke miopatije. Miopatija kod hipertireoze je prilično rijetka komplikacija. Karakterizira ga ptoza, bilateralna pareza mišića lica i mišića proksimalnih udova. U isto vrijeme, neki simptomi hipertireoze mogu biti maskirani slabošću mišića, ali tahikardija, pojačano znojenje i pojačano Štitnjača. Tetivni refleksi, za razliku od mnogih drugih oblika miopatije, ostaju normalni. Nakon korekcije hipertireoze mišićna slabost postupno nestaje.
Miopatija u hipotireozi. Hipotireoza kod dojenčadi može biti povezana sa slabošću mišića i hipotenzijom. U starije djece s miksedemom usporavaju se mišićne kontrakcije i opuštanje, u nekim slučajevima dolazi do hipertrofije mišića (Debre-Semelen sindrom). Kombinacija znakova kao što su mišićna slabost i hipertrofija ukazuje na mišićnu distrofiju.
Miopatija tijekom liječenja kortikosteroidima. Može zakomplicirati Itsenko-Cushingovu bolest, ali se češće razvija u liječenju velikim dozama sintetičkih steroida. Slabost je posebno izražena u mišićima zdjeličnog pojasa, što se očituje gegajućim (pačjim) hodom, otežanim penjanjem uz stepenice i pokušajima ustajanja iz sjedećeg položaja. Trzaj koljena je odsutan. Može doći do stanjivanja mišića. Miopatske promjene u mišićnom tkivu obično su beznačajne čak i uz jaku slabost. snaga mišića nakon ukidanja kortikosteroida polako se oporavlja (unutar nekoliko mjeseci).
Miopatija u hiperparatireoidizmu. Hiperparatireoidizam može biti povezan sa slabošću i hiporefleksijom zbog hiperkalemije. Obično nestaju brzo nakon paratiroidektomije.
Nedostatak karnitina (lipidna miopatija) prati nakupljanje velikih količina lipida u mišićima i, kao rezultat, kršenje opskrbe energijom potonjeg. Karnitin je jedna od bitnih komponenti sustava koji osigurava prijenos masnih kiselina iz dugi lanac iz citosola u mitohondrije, gdje se podvrgavaju oksidaciji. Slabost mišića razvija se u dva oblika nedostatka karnitina.
Nedostatak karnitina u mišićima klinički se očituje progresivnom slabošću njihovih proksimalnih skupina, češće u školske djece i adolescenata. Ponekad slabost povremena i u kombinaciji s mioglobinurijom. U teškim slučajevima može doći do paralize dišnih mišića. Povećane su razine enzima (kreatin kinaze i aldolaze) u serumu. Elektromiogram otkriva nespecifične promjene karakteristične za miopatiju. U biopsiji mišića možete vidjeti veliki broj kapljica masti. Razina karnitina u serumu se ne mijenja, ali u mišićima se smanjuje. Prepoznavanje patologije je bitno jer se ona može izliječiti. Često se pogrešno smatra mišićnom distrofijom. Učinak se može pojaviti nakon oralne primjene 100 mg/(kg/dan) karnitina. U nekim slučajevima liječenje kortikosteroidima je učinkovito.
- Sistemski nedostatak karnitina očituje se progresivnom miopatijom, uključujući kardiomiopatiju, i disfunkcijom jetre, praćenom klinikom jetrene encefalopatije poput Reyeovog sindroma. Nedostatak karnitina razlikuje se od potonjeg po ponavljajućem tijeku i izraženoj mišićnoj slabosti koja traje između razdoblja egzacerbacije encefalopatije. Razina kreatin fosfokinaze u serumu je izrazito povećana, količina karnitina je smanjena kako u serumu tako iu mišićima. Promjene u biopsiji slične su onima kod nedostatka karnitina u mišićnom tkivu. Slične kliničke i morfološke promjene, uključujući nedostatak karnitina, mogu se otkriti u kršenju metabolizma organskih kiselina, na primjer, kod metilmalonske i glutarne acidurije (sekundarni nedostatak karnitina).
Riža. 21-6 (prikaz, ostalo). Dijete s urođenim nedostatkom lijevog prsnog mišića. 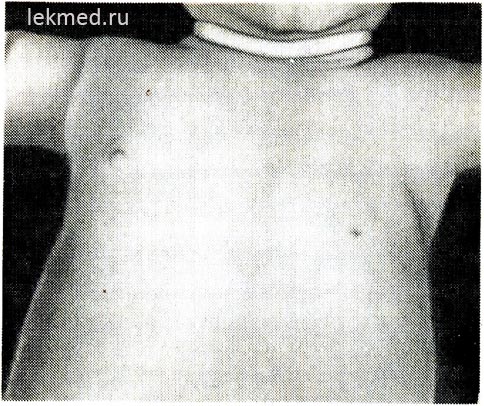
Obratite pozornost na odsutnost prednjeg aksilarnog nabora i niske bradavice.
Liječenje se sastoji u održavanju bolesnika na dijeti bogatoj ugljikohidratima i malo masti te uzimanju karnitina u dnevnoj dozi od 100 mg/kg.
Urođeni defekti mišića. Urođeni nedostatak mišića. Nerazvijenost mišića može biti vrlo česta i dovesti do potpune blokade pokreta zgloba ili kongenitalne artrogripoze. Kao urođena mana najčešće nedostaje jedan mišić. Prilično česta anomalija je odsutnost sternalnog dijela velikog prsnog mišića (slika 21-6), u nekim slučajevima ovaj se nedostatak kombinira sa sindaktilijom na zahvaćenoj strani (Polandov sindrom). Nedostatak prsnog mišića često prati mišićnu distrofiju. Kongenitalna odsutnost trbušnih mišića trbuha često je povezana s nedostacima u razvoju mokraćnog trakta.
Riža. 21-7 (prikaz, ostalo). Deformitet vrata i asimetrija lica u dječaka s kongenitalnim tortikolisom, neliječenim od 12. godine. 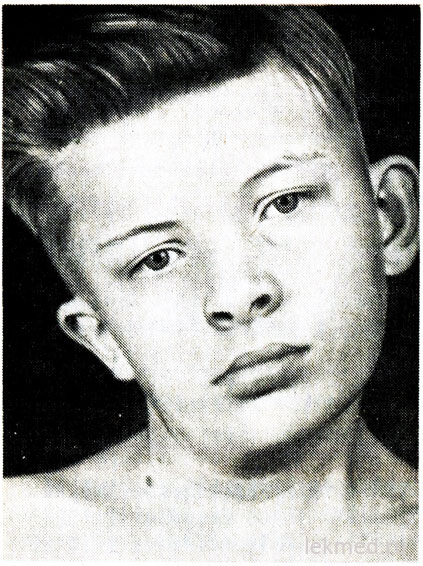
Kongenitalni tortikolis nastaje jednostranim skraćenjem ili kontrakturom sternokleidomastoidnog mišića. Glava bolesnika je nagnuta prema kontrakturi, a brada je usmjerena prema dolje u suprotnom smjeru (slika 21-7). Pri pokušaju ispravljanja položaja glave osjeća se značajan otpor mišića. U zahvaćenom mišiću palpiraju se područja zbijanja. Uzrok kvara je nejasan, dugo se smatrao rezultatom porodna ozljeda. Međutim, tortikolis se javlja kod djece rođene kirurškim zahvatom. carski rez; to sugerira da se u nekim slučajevima uzrok defekta odnosi na intrauterino razdoblje. Tortikolis treba razlikovati od patološkog naginjanja glave zbog deformacije vratnih kralješaka, kao što je Klippel-Weilova anomalija, te od prijeloma ili dislokacija vratnih kralješaka. Rentgenskim pregledom se isključuju. U starije djece, naginjanje glave može biti posljedica strabizma, distonije, tumora stražnje lubanjske jame i cervikalni leđne moždine, myositis ossificans, cervikalni limfadenitis odn dijafragmalna kila. U većini slučajeva, kongenitalni tortikolis može se ispraviti s terapeutska gimnastika. Međutim, kada kronični oblik tortikolis rezultira asimetričnim razvojem lica i glave (vidi sl. 21-7), što može zahtijevati disekciju mišića u kozmetičke svrhe.
kongenitalne miopatije. U ovu skupinu spada nekoliko rijetkih oblika nasljednih bolesti kod kojih se mišićna slabost i hipotonija javljaju već u djetinjstvu (vidi tablicu 22-1). Njihova točna dijagnoza je veliki značaj u smislu prognoze. Općenito, povoljna je za normalnu životnu aktivnost i životni vijek, za razliku od Werdnig-Hoffmannove bolesti ili kongenitalne mišićne distrofije. Biopsija mišića obično pomaže identificirati kongenitalne miopatije.
- Bolest središnje jezgre. Središnji dio mišićnih vlakana obojen je abnormalno, ali jednolično. Elektronski mikroskopski pregled otkriva smanjenje broja mitohondrija i depleciju sarkoplazmatskog retikuluma u središnjem dijelu vlakana.
Nemalinska miopatija. Izraz "ne grimizni" objašnjava se činjenicom da su u mišićnim vlaknima određene strukture poput niti.
Riža. 21-8 (prikaz, ostalo). Miotonična kontrakcija jezika (a) oštrim udarcem perkusijskog čekića po njegovoj desnoj polovici i kapcima (b) u djeteta s hiperkalijemijskim oblikom obiteljske periodične paralize. 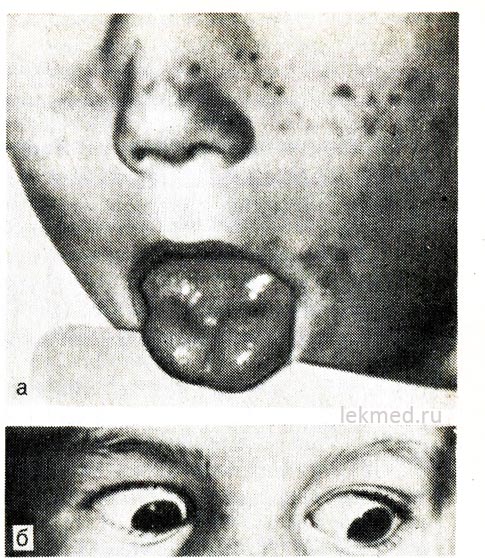
Kada gledate prema dolje, kapak ostaje skupljen.
Podaci elektronskog mikroskopa pokazuju da je to rezultat promjena u Z-pojasima miofibrila.
Mitohondrijske miopatije. Zabilježeni su neki oblici miopatija kod kojih se najvažnije promjene događaju u mitohondrijima mišićnih vlakana. Mogu se značajno povećati iu broju iu veličini. Slabost mišića i hipotonija mogu se utvrditi već u djetinjstvu, ali ponekad zamjetno napreduju tek u školi. Kardiomiopatija, encefalopatija i mliječna acidemija često prate ovu skupinu miopatija.
Miotonija. Ovo je stanje obilježje raznih mišićnih bolesti, kao što su myotonia dystrophica, hiperkalemijska obiteljska paroksizmalna paraliza i bolesti skladištenja glikogena. Miotonija se definira kao značajno kašnjenje u opuštanju mišića nakon voljnih ili prisilnih kontrakcija. Klinički se očituje nemogućnošću stiskanja šake ili vidljivom produljenom kontrakcijom mišića nakon njihova podražaja, koja se izražava oštrim nadražajem (sl. 21-8). To se može uočiti ako perkusijskim čekićem udarite površnu skupinu mišića, na primjer, mišiće jezika ili dlanove u području elevacije prvog prsta. Miotonija je potvrđena podacima elektromiografije. U ovom slučaju uočljiva je karakteristična spontana aktivnost mišića nakon njihovog opuštanja ili voljne kontrakcije (miotonična pražnjenja).
Myotonia congenita (Thomsenova bolest). Jedini znak ove bolesti, naslijeđen dominantnim tipom, je miotonija. Može se manifestirati u djetinjstvu u obliku usporenog gutanja i povraćanja
učinak nemogućnosti normalnog opuštanja mišića ždrijela. U starijem djetinjstvo miotonija se očituje kao nemogućnost bolesnika da otpusti prste stisnute u šaku. Pri prvom pokušaju da izvede neku vrstu pokreta mišići djeteta postaju tvrdi. Ponovljenim ponavljanjem istog pokreta, oni se donekle opuštaju. Tako npr. bolesno dijete ima velike poteškoće na početku prohodanja. Prvih nekoliko koraka obično čini vrlo oklijevajući i polako. Nakon nekoliko sekundi hod postaje normalan ili gotovo normalan. Simptomi miotonije pogoršani su nepovoljnim emocionalnim stanjem bolesnika i hlađenjem tijela. Snaga mišića ostaje normalna, mišići su dovoljno razvijeni i često zamjetno povećani, što stvara pogrešan dojam o sportskoj konstituciji bolesnika.
Dijagnoza se postavlja na temelju kliničke slike i podataka elektromiografije. Aktivnost enzima u serumu je u granicama normale. Jedini histološki znak je hipertrofija mišićnih vlakana.
Bolest se razlikuje od distrofične miotonije odsutnošću mišićne slabosti i atrofije te distrofičnih promjena u biopsiji mišićnog tkiva. Liječenje novokainom ili kinidin sulfatom prati učinak i indicirano je za funkcionalne poremećaje. Tijek bolesti je obično dobroćudan, a stanje bolesnika može se poboljšati s godinama.
paroksizmalna paraliza. Ovu skupinu bolesti karakterizira periodična slabost mišića s potpunim ili gotovo potpunim vraćanjem snage mišića u razdoblju između napada. Također uključuje nedostatak mišićne fosforilaze (McArdleova bolest).
Hiperkalijemijska paroksizmalna paraliza. Nasljedna epizodna adinamija ili paramiotonija prenosi se prema dominantnom tipu i posebno je teška u muškaraca. Obično počinje u ranom djetinjstvu (ponekad u djetinjstvu). Napadi se javljaju tijekom odmora nakon teškog mišićnog opterećenja. Slabost se razvija brzo i može trajati nekoliko sati. Posebno se osjeća u nogama; respiratorna funkcija obično nije oštećena. Često je adinamija popraćena miotonijom, koja traje između napada, što se najjasnije očituje u obliku kašnjenja u kretanju kapaka kada se gleda prema dolje (vidi sl. 21-8, b).
Razine kalija u serumu često su povišene tijekom napadaja, ali mogu biti potrebne višestruke studije tijekom nekoliko napada kako bi se to sa sigurnošću utvrdilo. Moguće je umjetno izazvati napadaj uz pomoć opterećenja kalijem (2-3 g oralno), ali to treba provoditi samo pod kontrolom EKG-a. Ponovljeni napadi zaustavljaju se dijakarbom. Teški oblici bolesti karakterizirani su razvojem kronične, blage slabosti i degenerativnih promjena u mišićima.
Hipokalemijska paroksizmalna paraliza. Obiteljska paroksizmalna paraliza, također naslijeđena prema dominantnom tipu, posebno je teška kod dječaka. Za razliku od hiperkalemijskog oblika, prvi napadaj javlja se u kasnom djetinjstvu ili ranoj adolescenciji. Razlog je obilan obrok bogat ugljikohidratima ili odmor nakon tjelesna aktivnost. Napad obično počinje na sljedeće jutro nakon teškog fizičkog napora i obilnog obroka. Karakterizira ga slabost mišića i arefleksija. Respiratorna funkcija može biti oslabljena. Može se pridružiti aritmija, uključujući ventrikularnu ekstrasistolu i tahikardiju. Napadaji mogu trajati više od 24 sata.U paralitičkoj fazi obično se smanjuje razina kalija u serumu (2-3 mmol/l). Temeljni nedostatak je nepoznat. Bolesnici s ponavljajućim teškim napadajima razvijaju kroničnu mišićnu slabost i patološke promjene u mišićima. Liječenje tijekom napadaja sastoji se od uzimanja kalijevog klorida; njegova početna doza je 2-3 g. Diakarb pomaže smanjiti učestalost napadaja.
Paroksizmalna mioglobinurija (idiopatska mioglobinurija). Idiopatska mioglobinurija je heterogena skupina poremećaja kod kojih se napadaji paralize s mioglobinurijom javljaju spontano ili nakon intenzivnog vježbanja. Bolest se nasljeđuje dominantno, vezano za X kromosom. Mišići, najčešće potkoljenice i bedra, tijekom napadaja postaju bolni i natečeni. Mokraća postaje tamnocrvena ili smeđa boja. Mioglobinurija može uzrokovati nekrozu bubrežnih tubula, koja je fatalna zbog zatajenja bubrega.
Dijagnoza se potvrđuje otkrivanjem mioglobulina u mokraći. Pozitivan benzidinski test u odsutnosti eritrocita u urinu potvrđuje prisutnost mioglobina u njemu, osobito ako hemoglobin nije otkriven u serumu. Hemoglobin se određuje spektrofotometrijski. Paroksizmalnu mioglobinuriju treba razlikovati od McArdleove bolesti, nedostatka karnitin palmitiltransferaze i mioglobinurije nakon neuobičajenog napornog vježbanja ili ozljede mišića u zdrava osoba. Mioglobinurija nakon teške mišićne vježbe javlja se kod pseudohipertrofične mišićne distrofije (Duchenneova bolest).
Liječenje se sastoji od odmora u krevetu; provesti ako je potrebno umjetna ventilacija pluća. Kako bi se spriječilo zatajenje bubrega, potrebno je pacijentu propisati obilno piće.
Nedostatak karnitin palmitiltransferaze. S nedostatkom ovog enzima, poremećen je prijenos dugolančanih masnih kiselina u segmente mitohondrija, u kojima se odvija oksidacija i proizvodnja ketona. Nedostatak izoenzima tipa II nasljeđuje se recesivno. Zbog njegovog nedostatka dolazi do poremećaja ketogeneze u tkivima, uključujući mišiće i jetru. Prvi znakovi bolesti češće se javljaju kod djece školske i adolescencije. Sastoje se od ponovljenih epizoda boli u mišićima, slabosti i vrućice nakon vježbanja ili gladovanja. Mioglobinurija koja prati napadaje može dovesti do zatajenja bubrega. Post dovodi do hipoglikemije. Između napada djeca izgledaju zdrava. Bolest se mora razlikovati od drugih stanja praćenih periodičnom slabošću i mioglobinurijom. Metoda određivanja aktivnosti karnitin palmitil transferaze ima diferencijalno dijagnostičku vrijednost. Smanjuje se u mišićima i hepatičkim tkivima, leukociti i kultura fibroblasta. Prehrana bogata ugljikohidratima s malo masti može pomoći u smanjenju napadaja.
Mišićne distrofije. Ove anomalije pripadaju skupini obiteljskih bolesti praćenih degeneracijom mišićnih vlakana. Klasifikacija mišićnih distrofija temelji se na značajkama kao što su vrijeme početka, brzina progresije, raspodjela lezija po mišićnim skupinama i način nasljeđivanja.
Pseudohipertrofična mišićna distrofija. Dječja, ili Duchenneov oblik, najčešći je oblik mišićne distrofije; učestalost mu je 0,14 na 1000 djece. U klasičnom obliku javlja se samo u dječaka, a nasljeđe vezano uz X kromosom javlja se u otprilike 50% probanda. U drugim slučajevima, bolest je posljedica novih mutacija. Zabilježen je rijedak oblik mišićne distrofije, klinički identičan Duchenneovom obliku, ali naslijeđen recesivnim tipom s istom učestalošću bolesti u dječaka i djevojčica. Pouzdano dijagnosticiranje bolesti rijetko je moguće kod djeteta mlađeg od 3 godine. Anamneza obično pokazuje da je dijete imalo zaostatak u razvoju motoričke funkcije, kasno je počeo sjediti, hodati i trčati, što, naravno, govori više rani početak bolesti. Česti su gegajući (pačji) hod, poteškoće u penjanju uz stepenice, hipertrofija mišića potkoljenice. kliničke manifestacije. U nekim slučajevima, drugi mišići također su uključeni u proces, posebno deltoidni, brachioradialis i jezični mišići.
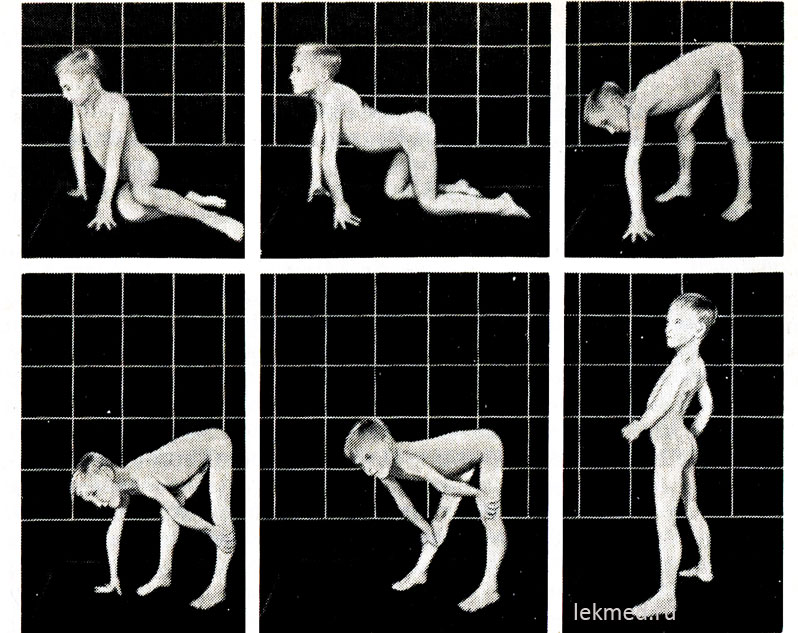
Riža. 21-9 (prikaz, ostalo). Tipični položaji pri ustajanju s poda (Goversov znak) u 7-godišnjeg djeteta s pseudohipertrofičnom miopatijom.
U stojećem stavu (zadnja slika) postoji značajno izražena lordoza.
U početku bolesti hipertrofirani mišići imaju značajnu snagu, ali kasnije ona opada (pseudohipertrofija), budući da povećanje mišićne mase nastaje zbog njihove masne infiltracije. Snaga hipertrofiranog gastrocnemius mišića značajno premašuje snagu mišića prednje površine noge, što objašnjava česte kontrakture kalkanealne tetive i hodanje djeteta na prstima. Slabost mišića zdjeličnog pojasa izražava se karakterističnim pačjim (gospodarskim) hodom i poteškoćama koje dijete doživljava kada ustane iz sjedećeg položaja na podu. S dovoljno teškim oblicima mišićne distrofije, dijete ima Goversov simptom: ustajući s poda, prvo klekne, oslanjajući se na ruke, a zatim se diže, uzastopno odgurujući ruke od potkoljenica, zglobovi koljena i bedra (sl. 21-9). Slabost mišića ramenog obruča možete utvrditi držeći dijete u povišenom položaju za pazuhe. Normalno, pokušava se držati pritiskom ruku na tijelo; s mišićnom distrofijom, čini se da klizi kroz ruke ispitivača. Bolesno dijete često ne može podići ruke iznad glave. U kasnijim fazama bolesti razvija se značajna atrofija mišića. Obično do 12. godine dijete više ne može hodati. Bolesnici u 75% slučajeva umiru prije navršene 20. godine života. Većina njih ima kardiomiopatiju, koja u nekim slučajevima uzrokuje iznenadna smrt. Ako je nasljeđe X-vezano, a bolest je započela u starijem djetinjstvu, očekivani životni vijek ostaje dug (Beckerova mišićna distrofija). Prosječni IQ za djecu s Duchenneom je 80; 25% djece ima mentalnu retardaciju.
Na diferencijalna dijagnoza Duchenneova mišićna distrofija trebala bi uključivati Werdnig-Hoffmannovu bolest u starije dojenčadi i bolesti mišića kao što su endokrine miopatije, nedostatak karnitina, bolesti skladištenja glikogena i polimiozitis. Ponekad se uz kontrakture kalkanealne tetive i hodanje djeteta na prstima može pretpostaviti cerebralna paraliza, ali kod mišićne distrofije nema karakterističnih znakova cerebralna paraliza spasticitet i hiperrefleksija.
Dijagnoza se temelji na određivanju aktivnosti enzima u serumu, podacima elektromiografije i biopsiji mišićnog tkiva. Aktivnost enzima, osobito kreatin-fosfokinaze, čak i prije razvoja klinički simptomičesto premašuje normu za 10 puta čak iu dojenčadi. Na elektromiogramu se prije svega otkriva smanjenje trajanja i smanjenje amplitude motoričkih potencijala. Histološke promjene sastoje se od degeneracije mišićnih vlakana. Često variraju u veličini i djelomično su zamijenjeni masnim i vezivnim tkivom. Veličina njihovih jezgri također varira. Dijagnoza se može postaviti pri rođenju određivanjem aktivnosti kreatin fosfokinaze. Metode za identifikaciju ženskih nositelja još nisu razvijene, unatoč činjenici da 60-80% njih pokazuje blagi ili umjereni porast njegove razine. Ovi su znakovi tipičniji za djetinjstvo nego za naredna razdoblja života.
učinkovite metode nema lijeka. Pacijent treba biti aktivan i sposoban hodati što je više moguće. Potrebno je osigurati da dijete izbjegava intenzivnu tjelesnu aktivnost jer može doći do pucanja mišićnih vlakana. U nekim slučajevima kirurško produljenje kalkanealne tetive poboljšava sposobnost hodanja, ali produljeno mirovanje nakon ortopedske korekcije može se povećati atrofija mišića. Genetsko savjetovanje ima važnu ulogu.
Kongenitalna mišićna distrofija. Bolest se nasljeđuje autosomno recesivno i karakterizirana je hipotenzijom mišića i slabošću u dojenčeta. Uvršten je u skupinu stanja definiranih kao "tromo dijete" (vidi tablicu 21-1). Početak bolesti odnosi se na intrauterini period. Ponekad novorođenče ima izraženu atrofiju mišića, njihove kontrakture, ograničenu pokretljivost zglobova. Razlikovanje od Werdnig-Hoffmannove bolesti je teško. Fascikulacije jezika, karakteristične za potonje, odsutne su kod mišićne distrofije. Tetivni refleksi su depresivni, ali nisu potpuno izgubljeni. Proces uključuje mišiće uključene u disanje, uključujući dijafragmu. U teškim slučajevima smrt nastupa prije navršene 1. godine života zbog zatajenja disanja; kod blažih oblika dugo se održava normalna vitalnost. Nije zabilježeno povećanje aktivnosti serumskih enzima, iako se javljaju distrofične promjene u mišićima.
Rame-facijalni oblik mišićne distrofije. Ovo je dovoljno blagi oblik mišićna distrofija se nasljeđuje autosomno dominantno. Obično počinje u dobi od 10-20 godina, a karakterizirana je slabošću i atrofijom mišića lica i ramenog obruča. Lice je potpuno amimično, bolesnik ne može zatvoriti oči i zazviždati. Bolest napreduje sporo i kompatibilna je s normalnim životnim vijekom. Dijagnoza se postavlja na temelju kliničke slike i vrste nasljeđa. Rezultati biopsije mišićnog tkiva ukazuju na distrofične promjene u njemu. Razine kreatin fosfokinaze u serumu mogu ostati normalne ili blago povišene.
Zdjelični oblik mišićne distrofije. Ovu skupinu heterogenih poremećaja karakterizira polagana progresija mišićne distrofije i nasljeđuje se autosomno recesivno. Početak bolesti odnosi se na starije djetinjstvo, adolescenciju ili odraslu dob. Obično su zahvaćeni mišići zdjeličnog pojasa.
Očni oblik miopatije. Distrofične promjene javljaju se uglavnom u vanjskim očnim mišićima. Bolest počinje u djetinjstvu ili adolescenciji. Uz to napreduje ptoza i ograničenje pokreta. očne jabučice. Ponekad se slabost proteže na mišiće lica i vrata. Bolest treba razlikovati od miastenije gravis i paralize kranijalnih živaca s tumorima moždanog debla.
Progresivna oftalmoplegija, koja počinje u djetinjstvu ili adolescenciji, povezana je s atipičnim pigmentnim retinitisom i srčanim blokom (Kearns-Sayersov sindrom). Obično je povezana s progresivnom ataksijom, usporenim rastom i pubertetom. Pod sarkolemom mišića utvrđuju se velike nakupine atipičnih mitohondrija. Genetska priroda ovog procesa nije utvrđena. S pacemakerom možete kontrolirati mogućnost iznenadne smrti od poremećaja srčanog provođenja.
Miotonična distrofija. Unatoč činjenici da miotonična distrofija počinje kao kod odrasle osobe, njezin se početak sve češće bilježi kod dojenčadi, a kasnije i djece. Nasljeđuje se autosomno dominantno. Njegov početak u djetinjstvu ukazuje na to da majka pati od miotonije. U skladu s tim, intrauterini čimbenici mogu utjecati na težinu bolesti kod djeteta. Već u trenutku rođenja može odrediti mišićna hipotenzija nedostaje mu sposobnost sisanja. Zaostajanje u tjelesnom i mentalnom razvoju obično se otkriva kasnije. U ranom djetinjstvu mišićna slabost i atrofija šire se uglavnom na mišiće lica, čeljusti i temporalne mišiće. Obično se primjećuje bilateralna ptoza. Dijagnostički značajne metode uključuju perkusiju mišića, elektromiografiju; Tipična za ove bolesnike je nemogućnost otpuštanja ruke stisnute u šaku (vidi Kongenitalna miotonija). Slabost i atrofija mišića udova i zdjeličnog pojasa (obično distalnih skupina) otkrivaju se u starijem djetinjstvu ili adolescenciji. U odraslih, ova bolest prati katarakta, ćelavost, atrofija testisa.
Dijagnoza se temelji na identifikaciji znakova miotonije, karakterističnoj raspodjeli mišićne slabosti, nasljeđivanju prema dominantnom tipu i distrofičnim promjenama u mišićima. U djetinjstvu tijek bolesti može biti nepovoljan, često praćen mentalna retardacija. Do adolescencije slabost mišića dolazi do izražaja. S funkcionalnim poremećajima indicirano je liječenje novokainom i kinidinom.

