DEDNE BOLEZNI ŽIVČEVJA
PREDAVANJE 16
Degenerativne bolezni s prevladujočo lezijo živčno-mišičnega aparata so največja skupina vseh dednih bolezni.
Izjemno pomembni in pogosto odločilni pri diagnostiki živčno-mišičnih bolezni so rezultati elektrofizioloških in biokemične raziskave. Enako velik je pomen patomorfoloških izvidov. Študija mišične biopsije pod svetlobnim mikroskopom pomaga razlikovati med miogeno in nevrogeno atrofijo. Histokemična preiskava je potrebna za odkrivanje presnovnih mišičnih lezij in elektronska mikroskopija odkril cel velik razred bolezni - neprogresivne miopatije.
Nežno raztezanje prizadetih sklepov zmanjša nastanek kontraktur in tako vodi do počasnejšega napredovanja bolezni. V tem primeru se izvajajo različne specifične vaje, tlačni povoj Urias pa se je izkazal za učinkovitega pri okvarjeni globini uda in se ob odpustu pogosto šteje za domačo zdravljenje. Senzomotorična vadba bolnikov z mišičnimi obolenji se izvaja v individualni ali skupinski terapiji in se osredotoča na koordinacijo in fino motoriko.
Zelo pomembna metoda Zdravljenje ljudi z mišičnimi boleznimi je ponovna vzpostavitev sposobnosti za vožnjo avtomobila. V ta namen poteka sodelovanje z zunanjo šolo vožnje na oddelku za delovno terapijo in vožnja v Bad Wildungnu se izvaja v ustreznem invalidskem vozilu z usposobljenim inštruktorjem vožnje.
Progresivne mišične distrofije. Izraz mišične distrofije je skupina genetsko pogojenih motenj, za katere je značilno napredovanje degenerativne spremembe v mišičnih vlaknih brez primarne patologije perifernega (spodnjega) motoričnega nevrona.
Različne oblike se med seboj razlikujejo po vrstah dedovanja, času začetka procesa, naravi in hitrosti njegovega poteka, posebnosti topografije mišične atrofije, prisotnosti ali odsotnosti psevdohipertrofije in umikov tetiv in drugih. znaki.
Dodaten pristop pri rehabilitaciji mišičnih obolenj je ustrezno svetovanje. To ne velja samo za ortoze, ampak za vse druge pripomočki na domu, Ciljna pomoč invalidom z mišičnimi obolenji je izjemno uporabna, invalidski vozički so natančno odmerjeni in prilagojeni potrebam oseb z mišičnimi obolenji. Tuš stol, dvigalo za kad, invalidski vozički, klančine za invalidski vozički in drugi.
Oddelek za fizikalno terapijo uporablja različne postopke za sprostitev pacienta. Poleg tega lahko ta oddelek zdravljenja zmanjša pogosto manjše simptome bolečine pri bolnikih z mišičnimi motnjami. Logoped je še en oddelek rehabilitacije za zdravljenje mišičnih bolezni. Tukaj je treba postaviti tudi drugo diagnozo z laringoskopijo ali določeno rentgenske študije. V usposobljeni rehabilitacijski ambulanti za zdravljenje mišičnih bolezni je pomemben predpogoj celostni terapevtski pristop k tej bolezni.
Večina mišičnih distrofij je klinično dobro raziskanih natančen opis narejen konec prejšnjega stoletja. Toda kljub skoraj stoletni zgodovini preučevanja miodistrofije ostajajo vprašanja njihove patogeneze in zdravljenja še danes nerešena. Veliko upanja polagamo na molekularno genetiko, s pomočjo katere je že določena lokacija genov številnih nosoloških oblik.
Sem sodijo diplomirani študenti psihologije fakultete. Prizadete osebe trpijo zaradi telesne pomanjkljivosti in s tem povezanih posledic v zasebnem življenju - Pričakovana življenjska doba je pogosto povezana z depresivna razpoloženja in psihosomatskih težav. Pri tem je v veliko pomoč podpora individualnim pogovorom ali spoznavanje tehnik sproščanja, kot sta avtogeni trening ali progresivna mišična sprostitev.
Vprašanja v zvezi z zakonom o zdravstvena nega, huda invalidnost, delovno mesto, stik z delodajalcem za poklicno rehabilitacijo ali vključitev v podjetje je treba omeniti kot pogoste teme socialnega svetovanja. Poleg tega je pogosto ciljno svetovanje v posvetovanjih pogosto zastavljeno vprašanje za bolnike z mišičnimi motnjami. Končno načrt terapije med rehabilitacijo v Bad Wildungnu vključuje tudi koordinacijo kvalificiranega specialista ortopeda. Vicerek je glavni zdravnik ortopedskega oddelka klinike v Hombergu v Bad Wildungenu.
Diagnoza mišičnih distrofij pogosto predstavlja velike težave. Obstaja velika variabilnost kliničnih manifestacij, majhno število družinskih članov pa otežuje določitev vrste dedovanja.
Značilna motorična napaka pri bolnikih z mišičnimi distrofijami je "račja" hoja: bolnik hodi zibajoč se na eno stran. Povezan je predvsem s šibkostjo glutealnih mišic, predvsem srednjih in malih, ki fiksirajo medenico glede na stegnenico. Zaradi tega bolezen povzroči nagib medenice proti neoporni nogi (Trendelenburgov fenomen) in kompenzacijski nagib telesa v nasprotna stran(Duchennov fenomen). Pri hoji se stran klanca nenehno spreminja. Te spremembe lahko preverimo tudi s Trendelenburgovim testom, tako da bolnika prosimo, naj dvigne eno nogo in jo pokrči pod pravim kotom v kolenskem in kolčnem sklepu: medenica na strani dvignjene noge se spusti (in se ne dvigne kot običajno) zaradi šibkosti gluteus medius mišice oporne noge.
Klinika izvaja redna izobraževanja, pa tudi sodelovanje na ustreznih kongresih in simpozijih. To srečanje je potekalo zlasti. Pogosto pride tudi do povečanja mišičnih encimov. Bolezni se navadno razvijajo počasi in polzeče, pojavljajo pa se hitre bolezni. Če sumite, da imate miopatijo ali ste vpleteni v znano nevromuskularno bolezen, se lahko naročite.
Zdravljenje je odvisno od poseben razlog, vnetna mišična bolezen lahko na primer Na primer različna imunoterapevtska sredstva. Podroben pregled v tabelah genov nevromuskularnih bolezni je mogoče najti različne genetske mišične bolezni.
Ko se dvigne iz vodoravnega položaja, se bolnik s hudo mišično oslabelostjo proksimalnih mišic težko prevrne na trebuh, nato pa se z rokami nasloni na tla, se postavi na vse štiri in nato z rokami na golenicah, nato na bokih , se postopoma zravna. Ta pojav »izbiranja po svoje« se imenuje Gowersov manever. Pogosto je povezana s šibkostjo gluteus maximus mišic.
Zdravila, ki lahko povzročijo ali povečajo mišično bolezen
V naši ordinaciji so na voljo vse potrebne diagnostične možnosti. Pri tem je vključena praksa, organizirana so redna izobraževanja s področja živčno-mišičnih bolezni. Prizadete osebe se lahko vpišejo tudi v register bolnikov. Registri bolnikov so pomemben prvi korak k boljšemu razumevanju raziskav razvoja bolezni in terapije.
Poškodbe srčne mišice
V tem je pregledana možna vpletenost srčne mišice v miopatijo in druge nevromuskularne bolezni.Duchennova miodistrofija. Psevdohipertrofična Duchennova mišična distrofija se pojavlja pogosteje kot vse druge bolezni mišičnega sistema (30 na 100.000 živorojenih otrok). Zanjo je značilen zgodnji začetek in maligni potek. Klasična slika se kaže s spremembo hoje pri otroku v starosti 2-5 let, pri 8-10 letih otroci že težko hodijo, pri 14-15 letih so običajno popolnoma imobilizirani. Otroci imajo več zgodnja starost začetni simptomi se kažejo z zaostankom v motoričnem razvoju: pozneje začnejo hoditi, ne morejo teči in skakati. Bolniki umrejo v 2. ali 3. desetletju življenja.
Mislimo na vse živčno-mišične bolezni. Po Waltonovi klasifikaciji je 800 oblik. Ta simptom je lahko omejen na nekaj mišičnih skupin ali, odvisno od bolezni, lahko najdemo celotno muskulaturo. Izguba mišic, zmanjšanje mišična masa in mišična oslabelost so glavni simptomi nevromuskularnih bolezni. Zdravila se imenujejo zdravila za zmanjšanje atrofije mišično-skeletnih mišic. Vendar pa še ne zagotavljajo neposredne diagnoze. Obstaja več razlogov, ki lahko vodijo do delnega zmanjšanja mišične mase, ki je deloma povezana s celotnim telesom.
Eden od prvih znakov bolezni je zbijanje telečjih mišic in postopno povečanje njihovega volumna zaradi psevdohipertrofije. Atrofija mišic stegna, medeničnega pasu je pogosto prikrita z dobro razvitim podkožnim maščobnim tkivom. Postopoma gre proces navzgor in se razširi čez ramenski obroč, hrbtne mišice in nato na proksimalne dele rok.
Pravi vzrok takšne mišične atrofije ali mišične oslabelosti se lahko skriva v oddaljenih, povsem drugih delih telesa. Vzroki so lahko v živčnih celicah hrbtenjača, odgovoren za gibanje, v dovodnih živcih, ko se živec prenese na mišico, ali v mišicah samih.
Zdravljenje nevromuskularnih bolezni
Obstajajo obetavna zdravila za zdravljenje miozitisa, miastenije gravis in endokrine miopatije. Prvi pristopi k upočasnitvi so možni z lateralna skleroza amiotrofični. V primeru dednih mišičnih distrofij in spinalnih mišičnih atrofij vzročno zdravljenje še ni ugotovljeno. Posebna pričakovanja za prihodnost so namenjena gensko zdravljenje. Pri vseh živčno-mišičnih boleznih je simptomatika dosledna fizioterapevtska podpora v kombinaciji z ortopedskimi ukrepi, ponekod z respiratorno podporo, ponekod z možnostjo presaditve srca.
IN končni fazi mišična oslabelost se lahko razširi na mišice obraza, žrela, dihalnih mišic.
V napredovali fazi bolezni so značilni simptomi, kako " račja hoja”; poudarjena ledvena lordoza, pterygoid scapulae, simptom "ohlapnega ramenskega obroča". Značilne so zgodnje mišične kontrakture in retrakcije tetiv, zlasti Ahilove tetive. Kolenski refleksi izpadejo zgodaj, nato pa refleksi iz zgornjih okončin.
Zdravljenje vključuje tudi kompenzacijo telesnih omejitev z ustreznimi sredstvi. Nemško združenje za mišične bolezni je ustanovilo neodvisen virski center. Obstaja tudi možnost testiranja izdelkov doma v dveh standardnih stanovanjih ali preživeti le nekaj dni v stanovanjih brez ovir.
Mišične bolezni Neodvisne mišične bolezni so relativno redke. Po drugi strani pa se pogosto zgodi, da mišice prizadenejo druge osnovne bolezni, predvsem bolezni živčni sistem; nalezljive bolezni kot sta tifus in tuberkuloza; Parazitske bolezni, trinity in prašičja glista, pa tudi hormonske motnje.
Psevdohipertrofija se lahko razvije ne samo v gastrocnemius, ampak tudi v glutealnih, deltoidnih, trebušnih in jezikovnih mišicah. Zelo pogosto srčna mišica trpi zaradi vrste kardiomiopatije. Motnje v ritmu srčne aktivnosti, širjenje meja srca, gluhost tonov, spremembe EKG. Akutno srčno popuščanje je največ pogost vzrok smrti zaradi Duchennove miodistrofije. Pri obdukciji ugotovimo fibrozo in maščobno infiltracijo srčne mišice.
Enostavna izguba mišic se pogosto pojavi kot posledica ohranjanja ali počitka. Krčenje mišic lahko povzročijo adhezije ali poškodbe zaradi slabe cirkulacije - Kot posledica asociacij zadušitve. Vzrok za bolečine v mišicah je lahko mišična revma, preobremenjenost, prehlad, presnovne motnje ali preobremenjenost določenih mišičnih skupin zaradi deformacij skeleta.
miom - benigni tumor mišično tkivo. Polimiozitis: Mišična bolezen s podobnimi simptomi kot dermatomiozitis, vendar brez kožni izpuščaj. Glede vzroka se domneva, da se kršitev nanaša na avtoagresije, kot na trpljenje, ki nastane kot posledica napada telesa na lastne strukture.
Pogosto pride do motenj gibljivosti gastrointestinalnega trakta.
Zmanjšana inteligenca je pogost simptom. Zanimivo je dejstvo, da je v nekaterih družinah oligofrenija močno izražena, v drugih razmeroma zmerno. Sprememba višjih duševnih funkcij običajno ne napreduje in ni v korelaciji z resnostjo mišične okvare. Tega ni mogoče razložiti samo s pedagoško zanemarjenostjo bolnih otrok, ki zgodaj izključujejo otroške skupine, jih ne obiskujejo. vrtec in šolo zaradi gibalne oviranosti. CT in MRI pogosto odkrijeta cerebralno atrofijo, ki je verjetno povezana z okvarjenim prenatalnim razvojem možganov.
Obstaja več kot 200 oblik mišičnih bolezni, nekatere so poimenovane po svojih odkriteljih, druge pa po motnji. Najpogostejše so tri podskupine. Progresivna mišična distrofija Spinalna mišična atrofija Nevronska mišična atrofija. V klinični praksi so te bolezni redke in nujno potrebne v rokah nevrologa.
Genetska napaka v kromosomu 19 je lezija mišične celice ki jih še danes ni mogoče zdraviti. Razlog je verjetno sprememba membranskega sistema mišičnih celic. Mistična distrofija lahko napreduje do popolnega uničenja mišičnih celic. Najprej so prizadete mišice na obrazu, rokah, podlakteh, spodnjih nogah in stopalih. Bolezen se lahko pojavi pri moških in ženskah vseh starosti.
Pogosto se pri otrocih razvije adiposogenitalni sindrom, včasih drugi znaki endokrine insuficience. Pogosto najdemo spremembe v skeletnem sistemu: deformacijo stopal, prsni koš, hrbtenica, difuzna osteoporoza.
Posebnost Duchennova oblika je visoka stopnja hiperencimemija že na zgodnje faze razvoj procesa. Tako lahko raven encima, specifičnega za mišično tkivo - kreatinin fosfokinaze - v krvnem serumu preseže več deset in celo stokrat. normalno delovanje. Močno (10-100-krat) povečanje kreatinin fosfokinaze (CPK) pri nevromuskularni patologiji bi moralo spodbuditi razpravo predvsem o naslednjih boleznih: Duchennova bolezen, Beckerjeva bolezen, poliomiozitis in dermatomiozitis, paroksizmalna mioglobulinurija, distalna miodistrofija. Šele v napredovalih fazah bolezni se stopnja hiperencimemije postopoma zmanjšuje. Obstajajo poročila o povečanju CPK v fazi intrauterinega razvoja.
Zakasnjena mišična sprostitev je značilna po mišična napetost. Posledice so mišična oslabelost in omejitev gibanja v nogah, rokah in rokah, poslabšanje fine motorike. Nekatere boleče mišice so še posebej močne zaradi značilne sestave spodnjega tkiva mišična vlakna vgrajen v maščobno in vezivno tkivo.
Imenuje se tudi kot prizadetost hrbtenjače mišični krč. Obstaja do 30 različne oblike spinalna mišična atrofija. Najpogostejša oblika je proksimalna spinalna mišična atrofija, opisana tukaj. Označen je na naslednji način po začetku trupa.
Duchennova mišična distrofija se prenaša na X-vezan recesiven način. Gen se nahaja na kratkem kraku kromosoma X. Pogostost genske mutacije je precej visoka (30%), kar pojasnjuje veliko število sporadični primeri.
Mutacija (najpogosteje delecija) vodi do spolne ali skoraj popolne odsotnosti genskega produkta - strukturnega proteina distrofičnega. Fiziološka vloga distrofija ni v celoti ugotovljena. Najdemo ga v visokih koncentracijah v sarkolemi in očitno igra vlogo pri ohranjanju celovitosti te membrane. Odsotnost distrofije povzroča strukturne spremembe v sarkolemi, kar posledično vodi do izgube intracelularnih komponent in povečanega vnosa kalcija, kar na koncu vodi do smrti miofibril. Menijo, da je pomanjkanje distrofičnih v sinaptičnih conah kortikalnih nevronov vzrok za duševno zaostalost.
Razlog je verjetno genetska okvara. Živčne celice v hrbtenjači so okužene sprednje skvamozne celice. Domnevno je vzrok živčni sistem živcev. To velja le za motorični živčni sistem. Deli živčnega sistema, ki so odgovorni za občutek dotika, zaznavanje bolečine in temperature, ostanejo nedotaknjeni. funkcija Mehur in rektum se ne poslabša.
Oznaka predstavlja tudi izgubo mišic zaradi živcev. Vzrok je skoraj vedno genetska okvara, ovojnice živčnih vlaken se nenormalno zadebelijo ali pa se uničijo same živčne ovojnice. Vklopljeno živčna vlakna vpliva na roke in noge.
Za medicinsko genetsko svetovanje je zelo pomembna ugotovitev heterozigotnega nosilstva. Z Duchennovo miodistrofijo pri heterozigotih je v približno 70% primerov subklinična in včasih celo jasni znaki mišična patologija - nekaj zbijanja in celo povečanje telečjih mišic, hitra utrujenost mišic med fizičnim naporom, spremembe v EMG in patomorfološkem pregledu mišičnih biopsij. Najpogosteje heterozigotni nosilci kažejo povečanje aktivnosti kreatinin fosfokinaze.
Intenzivnost hitrosti prevodnosti živcev se upočasni. Začne se v spodnjih okončinah z izgubo mišic in sočasno mišično oslabelostjo. Simptomi se nato povečajo v spodnjih okončinah, kasneje pa prizadenejo roke in podlakti. Senzorične motnje so majhne. možne so avtonomne motnje, kot sta prekomerno ali premajhno potenje in motnje krvnega pretoka. Lahko se pojavijo manjši spastični simptomi v nogah.
Mišična oslabelost je posledica mišične oslabelosti, dokler mišice niso popolnoma funkcionalne, kar povzroči znatne resne omejitve gibanja v nogah, rokah in rokah. Mišice so zelo pomemben del našega telesa. Brez mišic telo izgubi sposobnost gibanja in izvajanja različnih dejanj. Pravzaprav brez človeka mišični sistem verjetno ne boš mogel preživeti. To je posledica dejstva, da večina organov v prebavni sistem sestavljajo mišice in tudi samo srce, ki črpa kri, je prav tako mišica.
Ob prisotnosti klinične slike Duchennove miodistrofije pri ženskah je treba najprej izključiti možnost anomalije na kromosomu X - Shereshevsky-Turnerjev sindrom (XO), Morrisov sindrom (XY) ali mozaicizem pri teh sindromih.
Duchennova mišična distrofija, ki se začne razvijati že v prenatalnem obdobju, je v bistvu prirojena miopatija in jo lahko diagnosticiramo kmalu po rojstvu z biopsijo mišice in določitvijo aktivnosti CPK.
Beckerjeva miodistrofija. Poleg hude, maligne oblike X-vezane Duchennove miodistrofije obstaja benigna oblika - Beckerjeva bolezen. Glede na klinične simptome je zelo podoben obliki Duchenne, vendar se praviloma začne pozneje - pri 10-15 letih, teče nežno, bolniki ostanejo sposobni delati dolgo časa, pri starosti 20 let. -30 let in pozneje še lahko hodijo. Plodnost ni zmanjšana, zato je bolezen včasih zaslediti v več generacijah družine: bolan človek prenese bolezen na svojega vnuka preko svoje hčere (»učinek dedka«). Začetni simptomi, tako kot pri Duchennovi bolezni, se kažejo v šibkosti mišic medeničnega obroča, nato v proksimalnih spodnjih okončinah. Bolniki spremenijo svojo hojo, imajo težave pri vzpenjanju po stopnicah, pri vstajanju z nizkega sedeža. Zanj je značilna psevdohipertrofija telečjih mišic. Retrakcija petne (Ahilove) tetive je manj izrazita kot pri Duchennovi bolezni.
Pri tej obliki ni intelektualnih motenj, kardiomiopatija je odsotna ali rahlo izražena.
Kot pri drugih miodistrofijah, povezanih z X, Beckerjeva oblika bistveno poveča aktivnost CPK, čeprav v manjši meri kot pri Duchennu, ne presega 5000 enot. Gen za Beckerjevo bolezen je tako kot Duchennova bolezen lokaliziran v kratkem kraku kromosoma X; verjetno sta oba lokusa tesno povezana ali alelna. V nasprotju z Duchennovo boleznijo, pri kateri distrofije praktično ni, se pri Beckerjevi bolezni sintetizira nenormalna distrofija. Razlike najdemo tudi pri mišični biopsiji. Pri Beckerjevi mišični distrofiji mišična vlakna običajno niso okrogla, hialinska vlakna, značilna za Duchennovo mišično distrofijo, so izjemno redka.
Landouzy-Dejerine miodistrofija (obrazno-ramenska miodistrofija). Bolezen se prenaša avtosomno dominantno z visoko penetracijo, vendar nekoliko spremenljivo izraženostjo. Pojavlja se veliko manj pogosto kot Duchennova miodistrofija (0,4 na 100 tisoč prebivalcev). Domneva se, da je gen za to bolezen lokaliziran na 4. kromosomu. Ženske zbolijo pogosteje kot moški (3:1), Telesna preobremenitev, intenzivni športi, pa tudi neracionalno fizioterapija lahko prispevajo k hujšemu poteku bolezni.
Miodistrofija Landouzy-Dejerine je trenutno relativno ugodna oblika mišične patologije. Začne se pri starosti približno 20 let, včasih kasneje. Vendar pa je v družinskih primerih bolezni, ko je mogoče slediti dinamiki mlajših članov družine, mogoče zaznati nekatere oslabelosti mišic, na primer mišic obraza, in v zgodnejši starosti. .
Mišična oslabelost in atrofija se najprej pojavita v mišicah obraza oz ramenski obroč. Postopoma se te motnje razširijo na mišice proksimalnih rok in nato na spodnjih okončin. V večini primerov so najprej prizadete mišice sprednje površine nog (z razvojem visečega stopala), nato pa mišice proksimalnih nog. Na vrhuncu bolezni so močno prizadete krožne mišice očesa in ust, pectoralis major, anterior serratus in spodnji deli trapezaste mišice, latissimus dorsi, biceps, triceps mišice ramena. značilnost videz bolniki: tipičen obraz miopat s "prečnim nasmehom" ("La Giocondin nasmeh"), protruzija Zgornja ustnica("tapirske ustnice"), izrazite pterigoidne lopatice, svojevrstna deformacija prsnega koša s sploščitvijo v anteroposteriorni smeri in rotacijo znotraj ramenskih sklepov. Pogosto obstaja asimetrija lezije, tudi znotraj ene same mišice (npr. orbicularis oculi). Opazimo lahko psevdohipertrofijo gastrocnemiusa, deltoidnih mišic in včasih obraznih mišic. Kontrakture in retrakcije so zmerno izražene. tetivni refleksi dolgo časa se ohranijo, vendar včasih upadejo v zgodnji fazi.
Znaki poškodbe srčne mišice so redki. Serumska encimska aktivnost je rahlo povečana in je lahko normalna. Intelekt ne trpi. Pričakovana življenjska doba se v večini primerov ne zmanjša. Zanimivo je dejstvo, da EMG pri miodistrofiji Landouzy-Dejerine pogosto ni povsem značilen za mišično raven lezije. Pri nekaterih bolnikih (članih iste družine) lahko opazimo zmanjšanje amplitude biopotencialov, interferenčno vrsto krivulje, pri drugih, nasprotno, zmanjšanje frekvence in hipersinhrone aktivnosti, včasih s tipičnim piketom ograjni ritem. Ne smemo pozabiti na spinalno različico, ki posnema bolezen Landouzy-Dejerine.
Erb-Rothova miodistrofija (miodistrofija okončin). Prenaša se avtosomno recesivno, oba spola sta enako prizadeta. Začetek bolezni se v večini primerov nanaša na sredino 2. desetletja življenja (14-16 let), opisujejo pa jo kot zgodnjo, psevdo-Duchennovo obliko, ko se prvi simptomi pojavijo pred 10. letom in bolezen je huda in pozna različica z nastopom po 30 letih.
Potek bolezni je lahko hiter ali počasnejši, v povprečju nastopi popolna invalidnost v 15-20 letih od pojava prvih simptomov. Miodistrofija se začne s poškodbo mišic medeničnega obroča in proksimalnih nog (Leiden-Mobiusova oblika) ali iz ramenskega obroča (Erbova oblika). V nekaterih primerih sta ramenski in medenični obroč prizadeta hkrati. Mišice hrbta in trebuha precej trpijo. Bolniki imajo značilno "račjo" hojo, težko vstanejo iz ležečega in sedečega položaja, poudarjeno ledvena lordoza. Mišice obraza v večini primerov niso prizadete. Za to obliko so kontrakture in psevdohipertrofija neznačilne. Lahko se pojavi terminalna atrofija in retrakcija tetive. Inteligenca je običajno ohranjena. Srčna mišica večinoma ni prizadeta. Raven encimov v krvnem serumu se praviloma poveča, vendar ne tako močno kot pri X-vezani miodistrofiji. Obstajajo znaki, da je pri moških raven CPK višja kot pri ženskah. Obstaja pomembna razlika v izražanju mutantnega gena pri različnih družinskih članih – skupaj s hudimi klinična slika lahko so razmeroma blagi in celo izbrisani klinični simptomi. Smrt običajno nastopi zaradi pljučnih zapletov.
Ker je klinika miodistrofije okončin še posebej pripravljena posnemati živčno-mišične bolezni drugačne narave, je potrebno zlasti v občasnih primerih in z pozen začetek bolezni, opravite temeljit klinični pregled za izključitev spinalne amiotrofije, polimiozitisa, presnovnih, endokrinih, toksičnih, zdravilnih, karcinomatoznih miopatij. V preteklosti je prišlo do očitne pretirane diagnoze te oblike mišične distrofije.
Zdravljenje mišičnih distrofij. Terapevtske možnosti za mišične distrofije so zelo omejene. Etiološki in patogenetsko zdravljenje praktično ne obstaja. Simptomatsko zdravljenje je namenjeno predvsem preprečevanju razvoja kontraktur, ohranjanju obstoječe mišične moči in po možnosti nekoliko zmanjšanju stopnje atrofije. Glavna naloga je čim bolj podaljšati obdobje, v katerem se bolnik lahko samostojno giblje, saj se v ležečem položaju hitro povečajo kontrakture, skolioza in dihalne motnje. Medicinski kompleks mora vključevati terapevtske vaje, masažo, ortopedske ukrepe, zdravljenje z zdravili.
Terapevtska gimnastika je sestavljena iz pasivnih in aktivnih gibov v vseh sklepih v različnih položajih: stoje, sede, leže, z različnimi položaji okončin. aktivna gibanja Priporočljivo je izvajati v izometričnem načinu. Gimnastiko je treba izvajati redno večkrat na dan. Obenem velja opozoriti na pretirane obremenitve, zlasti tiste, ki jih spremlja preobremenitev mišic. Pomembne (zlasti po imobilizaciji pacienta) so dihalne vaje.
Ortopedski dogodki konzervativne (posebne opornice) in operativne narave (ahilotomija, transekcija gastrocnemius mišice), namenjene odpravljanju kontraktur in nastajajočih patoloških nastavitev okončin, so namenjene tudi ohranjanju možnosti samostojnega gibanja. V vsakem primeru je treba individualno pretehtati pričakovane koristi in morebitno škodo od kirurškega posega. Upoštevati je treba, da je pogosto (zlasti s hudo hiperlordozo in šibkostjo štiriglave stegenske mišice) ekvinovarusni položaj stopal kompenzacijskega pomena in po na primer ahilotomiji je lahko bolnik popolnoma imobiliziran. Pri razvoju kontraktur je priporočljivo skrbno raztegniti mišice do 20-30 krat na dan, čemur sledi opornica med spanjem.
Terapija z zdravili vključuje imenovanje presnovnih zdravil, namenjenih zapolnitvi pomanjkanja energije in beljakovin, vendar je njihova učinkovitost zelo dvomljiva. Uporabljajo se kalcijevi antagonisti (zaradi okvare, ugotovljene pri Duchennovi bolezni celične membrane kar vodi do povečanega vnosa kalcija v celico), imunomodulatorji, spojine, ki vsebujejo fosfor (ATP, fosfaden), vitamin E (100 mg peroralno 3-krat na dan). Dokazano je, da lahko uporaba prednizolona (0,75 mg / kg na dan) pri Duchennovi bolezni močno poveča mišično moč, vendar ta učinek traja največ eno leto in na splošno ne vpliva na izid bolezni. Zaradi resnih stranski učinki, ki nastane vri dolgotrajna uporaba zdravila, je njegova uporaba neustrezna. Ocene učinka anaboličnih steroidov sporna in njihovo imenovanje je pogosto povezano z neupravičenim tveganjem. Pri ocenjevanju učinka nekaterih zdravil po Duchennu je treba upoštevati, da lahko pri zmerni resnosti bolezni pri bolnikih, starih 3-6 let, pride do relativne stabilizacije stanja, povezanega s starostnim razvojem mišični sistem, pridobivanje motoričnih sposobnosti, ki lahko do neke mere začasno kompenzirajo tekoči distrofični proces.
Pomembna je korekcija bolnikove prehrane, priporočljiva je dieta z visoko vsebnostjo beljakovin in malo maščob ter zmanjšano kalorično vrednostjo z optimalno vsebnostjo vitaminov in mikroelementov. Pomembno vlogo ima psihološka podpora bolnika, nadaljevanje izobraževanja in pravilna poklicna orientacija.
Stran 44 od 44
Pri tem sodelujejo skeletne mišice patološki proces z različnimi degenerativnimi, presnovnimi in vnetne bolezni. V večini primerov pride do degeneracije mišičnih vlaken, pri kroničnih oblikah pa do njihove zamenjave. vezivnega tkiva in maščobe. Proksimalne mišične skupine so poškodovane močneje kot distalne, pa tudi spodnje okončine glede na zgornje. Bolnega otroka odlikuje tako imenovana račja hoja, ne more teči, plezati po stopnicah in vstati, če je v sedečem položaju. Njegovi tetivni refleksi so depresivni, stopnja njihovega izumrtja je sorazmerna s stopnjo oslabitve mišične moči. Občutljivost ni prizadeta.
Diagnostično dragocene laboratorijske metode vključujejo določanje aktivnosti encimov, zlasti kreatin fosfokinaze, v serumu. Ta encim, ki katalizira reakcijo: fosfokreatin + ADP-kreatin + ATP, je prisoten predvsem v možganskih celicah in mišičnem tkivu. Pri nekaterih difuznih mišičnih obolenjih, zlasti pri mišični distrofiji, njegove presežne količine prodrejo v medceličnino in kri. Pri bolnikih je aktivnost serumske laktat dehidrogenaze in glutamin oksalocetne transaminaze običajno povečana, vendar njihova široka porazdelitev v drugih tkivih, vključno z jetri, zmanjša specifičnost testa. Običajno je za razjasnitev diagnoze potrebna biopsija mišičnega tkiva.
Vnetne bolezni mišic. Vnetje mišičnega tkiva spremlja nekatere okužbe, zlasti trihinelozo, toksoplazmozo in tiste, ki jih povzroča virus Coxsackie. Pogosto je sestavni del kolagenskih bolezni, vključno z dermatomiozitisom, eritematoznim lupusom, nodoznim periarteritisom in revmatoidnim artritisom.
Polimiozitis. Difuzno izolirano vnetje mišic neznane etiologije se imenuje polimiozitis. Zanj je značilen hitro napredujoč potek, šibkost in bolečina v proksimalnih mišičnih skupinah. Pogosto so v proces vključene mišice vratu, zato je otroku težko dvigniti glavo in jo držati v tem položaju. Laboratorijski znaki mišičnega vnetja vključujejo povečanje ESR in število levkocitov. Vendar njihova odsotnost ne izključuje polimiozitisa. Ravni serumskih encimov so običajno povišane. Pri biopsiji mišic se določi degeneracija in delna regeneracija vlaken ter njihova infiltracija z limfoidnimi celicami. Težko je ločiti polimiozitis od mišične distrofije in dermatomiozitisa. Lahko predstavlja atipično obliko dermatomiozitisa, čeprav se histologija obeh stanj nekoliko razlikuje: za dermatomiozitis je značilen vaskulitis, ki ga pri polimiozitisu običajno ni. Pri slednjem je napoved nekoliko ugodnejša. Zdravljenje s kortikosteroidi spremlja učinek, vendar se lahko po njihovi prekinitvi pojavi ponovitev.
Progresivni osificirajoči miozitis. Etiologija te redke bolezni vezivnega tkiva in mišic ni znana. Poročajo, da za njo trpijo bratje in sestre, vključno z dvojčki, in se prenaša na krvne sorodnike v ravni liniji. Menijo, da je bolezen podedovana avtosomno dominantno. Fantje zbolijo 2-3 krat pogosteje kot dekleta.
Patološki znaki so odvisni od stopnje bolezni. V zgodnjih fazah najdemo lokalni edem in vnetne celične infiltrate v mišicah in kitah. Kasneje se območja vnetja nadomestijo z granulacijskim tkivom, sčasoma pa se v lezijah oblikujejo področja hrustanca in kostnega tkiva.
Skoraj 75 % bolnih otrok ima prirojene okvare razvoj, največkrat nerazvitost prstov na rokah in ankiloza falang prvih prstov na nogah ter nerazvitost prvih prstov na rokah, polidaktilija, ukrivljenost prstov na rokah, sindaktilija (noge), deformacija ušesne školjke gluhost, manjkajoči zobje. Enake prirojene malformacije so lahko pri sorodnikih bolnika, ki niso razvili progresivne bolezni vezivnega tkiva in mišic. Starost, pri kateri se lahko začne miozitis ossificans, se razlikuje od rojstva do starejšega otroštva. Običajno ločimo tri stopnje: 1) na mestih manjših lokalnih poškodb se pojavijo omejene, pogosto tople in mehke na dotik pastozne otekline mehkih tkiv; 2) po nekaj dneh simptomi vnetja izginejo in lezija se strdi; 3) pride do osifikacije prizadetega območja. Občasno se pojavijo nove lezije, predvsem na vratu in hrbtu. primarni simptom tortikolis lahko nastane, če se je proces razvil v sternokleidomastoidni mišici. Sčasoma se osifikacija razširi na številne kite in vezi. Nastopi ankiloza hrbtenice in sklepov rok in nog (slika 21-5). Vnetje se lahko razširi na temporomandibularne sklepe, kar oteži žvečenje. Kostni izrastki lahko štrlijo skozi kožo. V adolescenci bolezen pogosto povzroči popolno imobilizacijo in smrt zaradi odpoved dihanja in prenehanje dihanja, čeprav obstajajo poročila o primerih preživetja. Pri osificirajočem miozitisu obstaja veliko tveganje za nastanek osteogenega sarkoma.
riž. 21-5. Otrok s progresivnim osifikacijskim miozitisom (tipična drža z okorelostjo vratu in hrbta).
Včasih je patološki proces omejen na mesto predhodne poškodbe mehkih tkiv (miositis ossificans circumscripta). Razširjena kalcifikacija mišičnega tkiva se lahko pojavi tudi pri kroničnem polimiozitisu in dermatomiozitisu.
rezultate laboratorijske metodeŠtudije nimajo diagnostične vrednosti.
Serumske ravni kalcija, fosforja, alkalne fosfataze, pa tudi aktivnost kreatin fosfokinaze in drugih encimov ostajajo normalne. kosti v središču poškodbe se po strukturi ne razlikuje od norme.
Obstoječe metode zdravljenje ni zadovoljivo. V nekaterih primerih so pri uporabi ACTH in drugih kortikosteroidov opazili upočasnitev razvoja bolezni. Njihova vloga pri končnem rezultatu zdravljenja je vprašljiva.
Endokrine in presnovne miopatije. Miopatija pri hipertiroidizmu je dokaj redek zaplet. Zanj je značilna ptoza, dvostranska pareza obraznih mišic in mišic proksimalnih udov. Hkrati lahko nekatere simptome hipertiroidizma prikrije mišična oslabelost, vendar tahikardija, povečano potenje in povečano Ščitnica. Tetivni refleksi za razliko od mnogih drugih oblik miopatije ostanejo normalni. Po korekciji hipertiroidizma mišična oslabelost postopoma izgine.
Miopatija pri hipotiroidizmu. Hipotiroidizem pri dojenčkih je lahko povezan z mišično oslabelostjo in hipotenzijo. Pri starejših otrocih z miksedemom se mišične kontrakcije in sprostitev upočasnijo, v nekaterih primerih opazimo mišično hipertrofijo (Debre-Semelenov sindrom). Kombinacija znakov, kot sta mišična oslabelost in hipertrofija, kaže na mišično distrofijo.
Miopatija med zdravljenjem s kortikosteroidi. Lahko zaplete Itsenko-Cushingovo bolezen, vendar se pogosteje razvije pri zdravljenju velikih odmerkov sintetičnih steroidov. Oslabelost je še posebej opazna v mišicah medeničnega obroča, kar se kaže v vabljivi (račji) hoji, težkem vzpenjanju po stopnicah in poskusu vstajanja iz sedečega položaja. Trzanje kolena je odsotno. Lahko pride do redčenja mišic. Miopatske spremembe v mišičnem tkivu so običajno nepomembne tudi pri hudi šibkosti. moč mišic po ukinitvi kortikosteroidov se počasi (v nekaj mesecih) okreva.
Miopatija pri hiperparatiroidizmu. Hiperparatiroidizem je lahko povezan s šibkostjo in hiporefleksijo zaradi hiperkaliemije. Običajno hitro izginejo po paratiroidektomiji.
Pomanjkanje karnitina (lipidna miopatija) spremlja kopičenje velikih količin lipidov v mišicah in posledično moteno oskrbo slednjih z energijo. Karnitin je ena bistvenih sestavin sistema, ki zagotavlja prenos maščobnih kislin iz dolga veriga iz citosola v mitohondrije, kjer se oksidirajo. Mišična oslabelost se razvije v dveh oblikah pomanjkanja karnitina.
Pomanjkanje karnitina v mišicah se klinično kaže s progresivno šibkostjo njihovih proksimalnih skupin, pogosteje pri šolarjih in mladostnikih. Včasih je šibkost občasna in kombinirana z mioglobinurijo. V hujših primerih lahko pride do paralize dihalnih mišic. Serumske ravni encimov (kreatin kinaze in aldolaze) so povečane. Elektromiogram razkriva nespecifične spremembe, značilne za miopatijo. V mišični biopsiji lahko vidite veliko število kapljic maščobe. Nivo karnitina v serumu se ne spremeni, v mišicah pa se zmanjša. Prepoznavanje patologije je nujno, saj je ozdravljiva. Pogosto jo zamenjujejo z mišično distrofijo. Učinek se lahko pojavi po peroralni uporabi 100 mg / (kg / dan) karnitina. V nekaterih primerih je zdravljenje s kortikosteroidi učinkovito.
- Sistemsko pomanjkanje karnitina se kaže s progresivno miopatijo, vključno s kardiomiopatijo, in disfunkcijo jeter, ki jo spremlja klinika jetrne encefalopatije, kot je Reyejev sindrom. Pomanjkanje karnitina se od slednjega razlikuje po ponavljajočem se poteku in izraziti mišični oslabelosti, ki vztraja med obdobji poslabšanja encefalopatije. Raven kreatin fosfokinaze v serumu je izrazito povečana, količina karnitina je zmanjšana tako v serumu kot v mišicah. Spremembe v biopsiji so podobne tistim pri pomanjkanju karnitina v mišičnem tkivu. Podobne klinične in morfološke spremembe, vključno s pomanjkanjem karnitina, lahko odkrijemo pri motnjah presnove organskih kislin, na primer pri metilmalonski in glutarni aciduriji (sekundarna pomanjkljivost karnitina).
riž. 21-6. Otrok s prirojeno odsotnostjo leve velike prsne mišice. 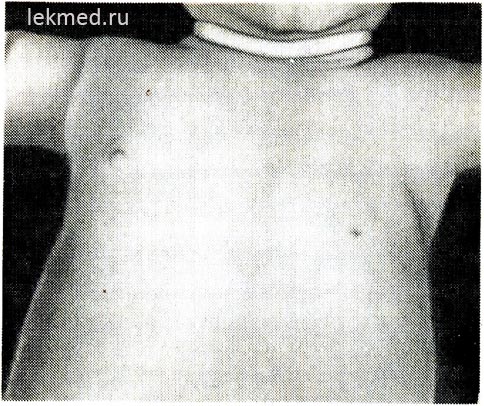
Upoštevajte odsotnost sprednje aksilarne gube in nizko ležeče bradavice.
Zdravljenje je sestavljeno iz vzdrževanja bolnika na dieti, bogati z ogljikovimi hidrati in nizko vsebnostjo maščob, ter jemanjem karnitina v dnevnem odmerku 100 mg / kg.
Prirojene okvare mišic. Prirojena odsotnost mišic. Mišična nerazvitost je lahko precej pogosta in povzroči popolno blokado sklepnih gibov ali prirojeno artrogripozo. Kot prirojena napaka najpogosteje manjka ena mišica. Dokaj pogosta anomalija je odsotnost sternalnega dela velike prsne mišice (slika 21-6), v nekaterih primerih je ta napaka kombinirana s sindaktilijo na prizadeti strani (Polandov sindrom). Odsotnost prsne mišice pogosto spremlja mišično distrofijo. Prirojena odsotnost trebušnih mišic trebuha je pogosto povezana z napakami v razvoju urinarnega trakta.
riž. 21-7. Deformacija vratu in asimetrija obraza pri dečku s prirojenim tortikolisom, nezdravljenim od 12. leta starosti. 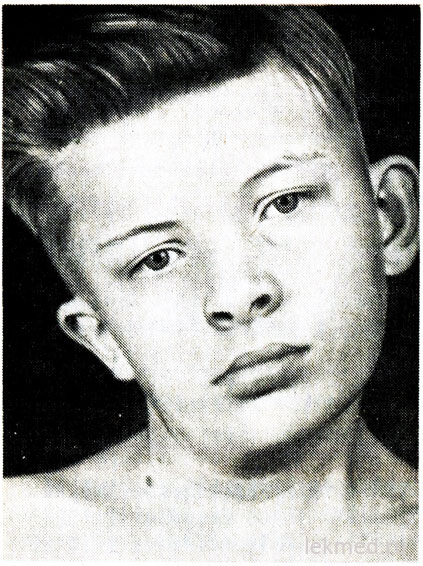
Prirojeni tortikolis je posledica enostranskega skrajšanja ali kontrakture sternokleidomastoidne mišice. Pacientova glava je nagnjena proti kontrakturi, brada pa je usmerjena navzdol v nasprotni smeri (slika 21-7). Ko poskušate popraviti položaj glave, se čuti pomemben mišični odpor. V prizadeti mišici se palpirajo območja zbijanja. Vzrok okvare ni jasen, dolgo časa je veljal za posledico porodna poškodba. Vendar se tortikolis pojavi pri otrocih, rojenih po operaciji. carski rez; to nakazuje, da se v nekaterih primerih vzrok okvare nanaša na intrauterino obdobje. Tortikolis je treba razlikovati od patološkega nagiba glave zaradi deformacije vratnih vretenc, kot je Klippel-Weilova anomalija, ter od zlomov ali izpahov vratnih vretenc. Z rentgenskim pregledom jih izključimo. Pri starejših otrocih je lahko nagnjena glava posledica strabizma, distonije, tumorjev zadnje lobanjske jame in materničnega vratu hrbtenjače, myositis ossificans, vratni limfadenitis oz diafragmalna kila. V večini primerov je mogoče prirojeni tortikolis popraviti z terapevtska gimnastika. Vendar, ko kronična oblika tortikolis povzroči asimetričen razvoj obraza in glave (glej sliko 21-7), kar lahko zahteva disekcijo mišice v kozmetične namene.
prirojene miopatije. V to skupino spada več redkih oblik dednih bolezni, pri katerih se mišična oslabelost in hipotonija pojavita že od otroštva (glej tabelo 22-1). Njihova natančna diagnoza je velik pomen v smislu napovedi. Na splošno je ugoden za normalno življenjsko aktivnost in pričakovano življenjsko dobo, v nasprotju z Werdnig-Hoffmannovo boleznijo ali prirojeno mišično distrofijo. Mišična biopsija običajno pomaga prepoznati prirojene miopatije.
- Bolezen osrednjega jedra. Osrednji del mišičnih vlaken je obarvan nenormalno, a enakomerno. Elektronski mikroskopski pregled razkrije zmanjšanje števila mitohondrijev in izčrpanost sarkoplazemskega retikuluma v osrednjem delu vlaken.
Nemalinska miopatija. Izraz "ne škrlatni" je razložen z dejstvom, da so v mišičnih vlaknih določene nitaste strukture.
riž. 21-8. Miotonična kontrakcija jezika (a) z ostrim udarcem z udarnim kladivom po desni polovici in vekah (b) pri otroku s hiperkalemično obliko družinske periodične paralize. 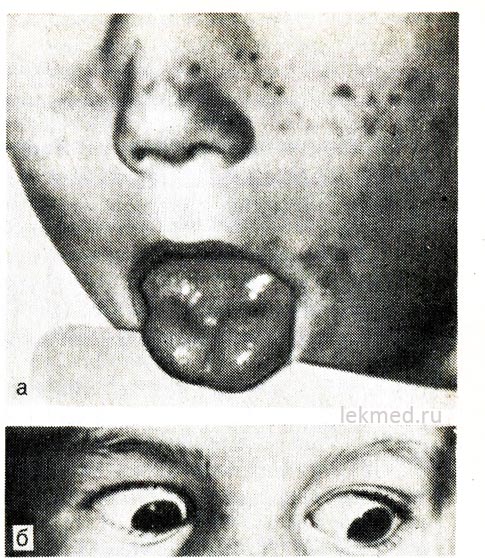
Pri pogledu navzdol veke ostanejo skrčene.
Podatki elektronske mikroskopske preiskave kažejo, da je to posledica sprememb v Z-trakovih miofibril.
Mitohondrijske miopatije. Poročali so o nekaterih oblikah miopatij, pri katerih se najpomembnejše spremembe pojavijo v mitohondrijih mišičnih vlaken. Lahko se opazno povečajo tako po številu kot po velikosti. Mišična oslabelost in hipotonija se lahko ugotovita že v otroštvu, včasih pa opazno napredujeta šele v šoli. To skupino miopatij pogosto spremljajo kardiomiopatija, encefalopatija in laktacidemija.
miotonija. To stanje je značilno za različne mišične bolezni, kot so myotonia dystrophica, hiperkalemična družinska paroksizmalna paraliza in bolezni shranjevanja glikogena. Miotonija je opredeljena kot znatna zamuda pri sprostitvi mišic po prostovoljnem ali prisilnem krčenju. Klinično se kaže v nezmožnosti stiskanja pesti ali v vidnem dolgotrajnem krčenju mišic po njihovi stimulaciji, ki se izraža v ostrem draženju (slika 21-8). To lahko opazimo, če z udarnim kladivom udarimo po površinski skupini mišic, na primer po mišicah jezika ali dlani v območju dviga prvega prsta. Miotonija je potrjena s podatki elektromiografije. V tem primeru je opazna značilna spontana aktivnost mišic po njihovi sprostitvi ali prostovoljni kontrakciji (miotonični izpusti).
Myotonia congenita (Thomsenova bolezen). Edini znak te bolezni, podedovan po dominantnem tipu, je miotonija. V otroštvu se lahko manifestira v obliki upočasnjenega požiranja in bruhanja, ki mu sledi
učinek nezmožnosti normalne sprostitve mišic žrela. V starejših otroštvo miotonija se kaže kot nezmožnost pacienta, da stisne prste, stisnjene v pest. Pri prvem poskusu izvajanja neke vrste giba postanejo otrokove mišice trde. Z večkratnim ponavljanjem istega giba se nekoliko sprostijo. Tako ima na primer bolan otrok velike težave na začetku hoje. Običajno prvih nekaj korakov naredi zelo obotavljajoče in počasi. Po nekaj sekundah hoja postane normalna ali skoraj normalna. Simptomi miotonije se poslabšajo z neugodnim čustvenim stanjem bolnika in ohlajanjem telesa. Mišična moč ostaja normalna, mišice so dovolj razvite in pogosto opazno povečane, kar ustvarja napačen vtis o športni konstituciji bolnika.
Diagnoza temelji na kliničnih izvidih in podatkih elektromiografije. Aktivnost encimov v serumu je v mejah normale. Edini histološki znak je hipertrofija mišičnih vlaken.
Bolezen se od distrofične miotonije razlikuje po odsotnosti mišične oslabelosti in atrofije ter distrofičnih sprememb v biopsiji mišičnega tkiva. Zdravljenje z novokainom ali kinidin sulfatom spremlja učinek in je indicirano za funkcionalne motnje. Potek bolezni je običajno benigen, bolnikovo stanje pa se s starostjo lahko izboljša.
paroksizmalna paraliza. Za to skupino bolezni je značilna periodična mišična oslabelost s popolno ali skoraj popolno obnovo mišične moči v obdobju med napadi. Vključuje tudi pomanjkanje mišične fosforilaze (McArdlova bolezen).
Hiperkalemična paroksizmalna paraliza. Dedna epizodna adinamija ali paramiotonija se prenaša po dominantnem tipu in je še posebej huda pri moških. Običajno se začne v zgodnjem otroštvu (včasih v povojih). Napadi se pojavijo med počitkom po težki mišični obremenitvi. Slabost se razvije hitro in lahko traja več ur. Še posebej se čuti v nogah; dihalna funkcija običajno ni oslabljena. Pogosto adinamijo spremlja miotonija, ki vztraja med napadi, kar se najbolj jasno kaže v obliki zakasnitve gibanja vek pri pogledu navzdol (glej sliko 21-8, b).
Raven kalija v serumu je med napadom pogosto povišana, vendar bo morda potrebnih več študij med več napadi, da se to z gotovostjo ugotovi. Možno je umetno izzvati napad s pomočjo obremenitve s kalijem (2-3 g peroralno), vendar ga je treba izvajati le pod nadzorom EKG. Ponavljajoče se napade ustavi diakarb. Za hude oblike bolezni je značilen razvoj kronične, blage oslabelosti in degenerativnih sprememb v mišicah.
Hipokalemična paroksizmalna paraliza. Družinska paroksizmalna paraliza, podedovana tudi po dominantnem tipu, je še posebej težka pri dečkih. V nasprotju s hiperkalemično obliko se prvi napad pojavi v poznem otroštvu ali zgodnji adolescenci. Razlog je uživanje obilnega obroka, bogatega z ogljikovimi hidrati, ali počitek po telesna aktivnost. Napad se običajno začne naslednje jutro po težkem fizičnem naporu in obilnem obroku. Zanjo je značilna mišična oslabelost in arefleksija. Dihalna funkcija je lahko oslabljena. Lahko se pridruži aritmija, vključno z ventrikularno ekstrasistolo in tahikardijo. Napadi lahko trajajo več kot 24 ur.V paralitični fazi se raven kalija v serumu običajno zniža (2-3 mmol / l). Osnovna napaka ni znana. Bolniki s ponavljajočimi se hudimi napadi razvijejo kronično mišično oslabelost in patološke spremembe v mišicah. Zdravljenje med napadi je sestavljeno iz jemanja kalijevega klorida; njegov začetni odmerek je 2-3 g Diakarb pomaga zmanjšati pogostost napadov.
Paroksizmalna mioglobinurija (idiopatska mioglobinurija). Idiopatska mioglobinurija je heterogena skupina motenj, pri katerih se napadi paralize z mioglobinurijo pojavijo spontano ali po intenzivni vadbi. Bolezen se deduje dominantno, vezano na kromosom X. Mišice, največkrat mečne in stegenske, med napadom postanejo boleče in otekle. Urin postane temno rdeč oz Rjave barve. Mioglobinurija lahko povzroči ledvično tubulno nekrozo, ki je usodna zaradi odpoved ledvic.
Diagnozo potrdimo z odkrivanjem mioglobulina v urinu. Pozitiven test z benzidinom v odsotnosti eritrocitov v urinu potrjuje prisotnost mioglobina v njem, še posebej, če hemoglobin ni zaznan v serumu. Hemoglobin se določi s spektrofotometrijo. Paroksizmalno mioglobinurijo je treba razlikovati od McArdlove bolezni, pomanjkanja karnitin palmitiltransferaze in mioglobinurije po neobičajni naporni vadbi ali poškodbi mišice. zdrava oseba. Mioglobinurija po težki mišični vadbi se pojavi pri psevdohipertrofični mišični distrofiji (Duchennova bolezen).
Zdravljenje je sestavljeno iz počitka v postelji; izvedite, če je potrebno umetno prezračevanje pljuča. Da bi preprečili odpoved ledvic, je treba bolniku predpisati obilno pijačo.
Pomanjkanje karnitin palmitiltransferaze. S pomanjkanjem tega encima je moten prenos dolgoverižnih maščobnih kislin v mitohondrijske segmente, v katerih poteka oksidacija in proizvodnja ketonov. Pomanjkanje izoencima tipa II se deduje recesivno. Zaradi njegovega pomanjkanja je motena ketogeneza v tkivih, vključno z mišicami in jetri. Prvi znaki bolezni se pogosteje pojavijo pri otrocih šole in adolescence. Sestavljeni so iz ponavljajočih se epizod mišične bolečine, šibkosti in vročine po vadbi ali postu. Mioglobinurija, ki spremlja napade, lahko povzroči odpoved ledvic. Postenje vodi v hipoglikemijo. Med napadi so otroci videti zdravi. Bolezen je treba razlikovati od drugih stanj, ki jih spremlja občasna šibkost in mioglobinurija. Metoda za določanje aktivnosti karnitin palmitil transferaze ima diferencialno diagnostično vrednost. Zmanjša se v mišicah in jetrna tkiva levkocitov in kulture fibroblastov. Uživanje prehrane, bogate z ogljikovimi hidrati in nizko vsebnostjo maščob, lahko pomaga zmanjšati napade.
Mišične distrofije. Te anomalije spadajo v skupino družinskih bolezni, ki jih spremlja degeneracija mišičnih vlaken. Razvrstitev mišičnih distrofij temelji na značilnostih, kot so čas nastanka, stopnja napredovanja, porazdelitev lezij po mišičnih skupinah in način dedovanja.
Psevdohipertrofična mišična distrofija. Otroška ali Duchennova oblika je najpogostejša oblika mišične distrofije; njegova pogostnost je 0,14 na 1000 otrok. V klasični obliki se pojavlja le pri dečkih, dedovanje, vezano na kromosom X, pa se pojavi pri približno 50 % probandov. V drugih primerih je bolezen posledica novih mutacij. Poročajo o redki obliki mišične distrofije, ki je klinično enaka Duchennovi obliki, vendar jo deduje recesivni tip z enako pogostnostjo bolezni pri dečkih in deklicah. Pri otroku, mlajšem od 3 let, je redko mogoče zanesljivo diagnosticirati bolezen. Anamneza običajno kaže, da je imel otrok zapozneli razvoj motorične funkcije, začel je pozno sedeti, hoditi teči, kar seveda kaže več zgodnji začetek bolezni. Pogosti so gatajoča (račja) hoja, težave pri vzpenjanju po stopnicah, hipertrofija mečnih mišic. klinične manifestacije. V nekaterih primerih so v proces vključene tudi druge mišice, zlasti deltoidne, brahioradialne in jezikovne mišice.
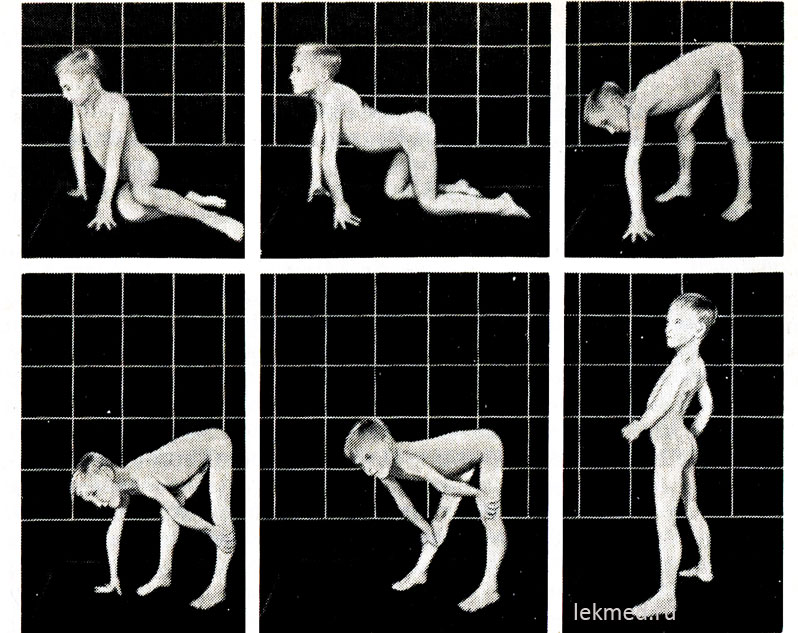
riž. 21-9. Tipični položaji pri vstajanju s tal (Goversov znak) pri 7-letnem otroku s psevdohipertrofično miopatijo.
V stoječem položaju (zadnja slika) je močno izražena lordoza.
Na začetku bolezni imajo hipertrofirane mišice znatno moč, kasneje pa se ta zmanjša (psevdohipertrofija), saj pride do povečanja mišične mase zaradi njihove maščobne infiltracije. Moč hipertrofirane gastrocnemius mišice znatno presega moč mišic sprednje površine noge, kar pojasnjuje pogoste kontrakture kalcanalne tetive in otrokovo hojo po prstih. Oslabelost mišic medeničnega obroča se izraža v značilni račji (gospodarski) hoji in težavah, ki jih ima otrok, ko vstane iz sedečega položaja na tleh. Pri dovolj hudih oblikah mišične distrofije ima otrok Goversov simptom: ko vstane s tal, najprej poklekne, se naslanja na roke, nato pa se dvigne, zaporedoma potisne roke iz golenice, kolenskih sklepov in stegna (slika 21-9). Šibkost mišic ramenskega obroča lahko ugotovite tako, da otroka držite v dvignjenem položaju za pazduhe. Običajno se poskuša držati tako, da roke pritisne na telo; pri mišični distrofiji se zdi, kot da polzi iz rok preizkuševalca. Bolan otrok pogosto ne more dvigniti rok nad glavo. V kasnejših fazah bolezni se razvije izrazita mišična atrofija. Običajno do 12. leta otrok ne more več hoditi. Bolniki v 75% primerov umrejo pred 20. letom starosti. Večina jih ima kardiomiopatijo, ki v nekaterih primerih povzroči nenadna smrt. Če je dedovanje X-vezano in se je bolezen začela v starejšem otroštvu, ostane pričakovana življenjska doba dolga (Beckerjeva mišična distrofija). Povprečni IQ za otroke z Duchenne je 80; 25 % otrok ima duševno zaostalost.
pri diferencialna diagnoza Duchennova mišična distrofija bi morala vključevati Werdnig-Hoffmannovo bolezen pri starejših dojenčkih in mišične bolezni, kot so endokrine miopatije, pomanjkanje karnitina, bolezni shranjevanja glikogena in polimiozitis. Včasih s kontrakturami kalcanealne tetive in otrokovo hojo po prstih lahko domnevamo cerebralno paralizo, vendar z mišično distrofijo ni značilnih znakov cerebralna paraliza spastičnost in hiperrefleksija.
Diagnoza temelji na določitvi encimske aktivnosti v serumu, podatkih elektromiografije in biopsiji mišičnega tkiva. Aktivnost encimov, zlasti kreatin fosfokinaze, še pred razvojem klinični simptomi pogosto presega normo za 10-krat tudi pri dojenčkih. Na elektromiogramu se najprej razkrije zmanjšanje trajanja in zmanjšanje amplitude motoričnih potencialov. Histološke spremembe so sestavljene iz degeneracije mišičnih vlaken. Pogosto so različno veliki in so delno nadomeščeni z maščobo in vezivnim tkivom. Tudi velikost njihovih jeder je različna. Diagnozo lahko postavimo ob rojstvu z določitvijo aktivnosti kreatin fosfokinaze. Metode za identifikacijo ženskih nosilcev še niso bile razvite, kljub dejstvu, da jih 60-80% kaže rahlo ali zmerno povečanje njegove ravni. Ti znaki so bolj značilni za otroštvo kot za naslednja življenjska obdobja.
učinkovite metode ni zdravila. Bolnik mora biti aktiven in čim več hoditi. Poskrbeti je treba, da se otrok izogiba intenzivni telesni dejavnosti, saj lahko povzroči pretrganje mišičnih vlaken. V nekaterih primerih kirurško podaljšanje petne tetive izboljša sposobnost hoje, vendar dolgotrajno počitek v postelji po ortopedski korekciji se lahko poveča mišična atrofija. Genetsko svetovanje ima pomembno vlogo.
Prirojena mišična distrofija. Bolezen se deduje avtosomno recesivno in je značilna mišična hipotenzija in šibkost pri dojenčku. Vključen je v skupino stanj, opredeljenih kot "počasen otrok" (glej tabelo 21-1). Začetek bolezni se nanaša na intrauterino obdobje. Včasih ima novorojenček izrazito atrofijo mišic, njihove kontrakture, omejeno gibljivost sklepov. Razlikovanje od Werdnig-Hoffmannove bolezni je težko. Fascikulacije jezika, značilne za slednje, so odsotne pri mišični distrofiji. Tetivni refleksi so oslabljeni, vendar niso popolnoma izgubljeni. Proces vključuje mišice, ki sodelujejo pri dihanju, vključno z diafragmo. V hujših primerih pride do smrti pred 1 letom starosti zaradi odpovedi dihanja; pri blažjih oblikah se normalna sposobnost preživetja ohranja dalj časa. Povečanja aktivnosti serumskih encimov ni opaziti, čeprav se v mišicah pojavijo distrofične spremembe.
Ramensko-obrazna oblika mišične distrofije. To je dovolj blaga oblika mišična distrofija se deduje avtosomno dominantno. Običajno se začne v starosti 10-20 let in je značilna šibkost in atrofija mišic obraza in ramenskega obroča. Obraz je popolnoma amimičen, bolnik ne more zapreti oči in zažvižgati. Bolezen napreduje počasi in je združljiva z normalno pričakovano življenjsko dobo. Diagnoza temelji na kliničnih izvidih in vrsti dedovanja. Rezultati biopsije mišičnega tkiva kažejo na distrofične spremembe v njem. Ravni serumske kreatin fosfokinaze lahko ostanejo normalne ali rahlo povišane.
Medenična oblika mišične distrofije. Za to skupino heterogenih bolezni je značilno počasno napredovanje mišične distrofije in se deduje avtosomno recesivno. Začetek bolezni se nanaša na starejše otroštvo, adolescenco ali odraslo dobo. Običajno so prizadete mišice medeničnega obroča.
Očesna oblika miopatije. Distrofične spremembe se pojavljajo predvsem v zunanjih očesnih mišicah. Bolezen se začne v otroštvu ali adolescenci. Z njim napredujeta ptoza in omejevanje gibov. zrkla. Včasih se šibkost razširi na mišice obraza in vratu. Bolezen je treba razlikovati od miastenije gravis in paralize kranialni živci s tumorji možganskega debla.
Progresivna oftalmoplegija, ki se začne v otroštvu ali adolescenci, je povezana z atipičnim pigmentnim retinitisom in srčnim blokom (Kearns-Sayersov sindrom). Običajno je povezana s progresivno ataksijo, zapoznelo rastjo in puberteto. Pod sarkolemo mišic se določijo velika kopičenja atipičnih mitohondrijev. Genetska narava tega procesa ni bila ugotovljena. S srčnim spodbujevalnikom lahko nadzorujete možnost nenadne smrti zaradi motenj srčnega prevajanja.
Miotonična distrofija. Kljub dejstvu, da se miotonična distrofija začne kot pri odraslem, se njen pojav vse pogosteje beleži pri dojenčkih in pozneje pri otrocih. Deduje se avtosomno dominantno. Njegov pojav v otroštvu kaže, da mati trpi za miotonijo. V skladu s tem lahko intrauterini dejavniki vplivajo na resnost bolezni pri otroku. Že ob rojstvu lahko določi mišična hipotenzija nima sposobnosti sesanja. Zaostanek v telesnem in duševnem razvoju se običajno odkrije pozneje. V zgodnjem otroštvu se mišična oslabelost in atrofija razširita predvsem na obrazne, čeljustne in temporalne mišice. Običajno opazimo dvostransko ptozo. Diagnostično pomembne metode vključujejo tolkala mišic, elektromiografijo; značilna za te bolnike je nezmožnost sprostitve roke, stisnjene v pest (glejte Prirojena miotonija). Slabost in atrofija mišic okončin in medeničnega obroča (običajno distalnih skupin) se odkrijejo v starejšem otroštvu ali adolescenci. Pri odraslih to bolezen spremljajo katarakta, plešavost, atrofija testisov.
Diagnoza temelji na identifikaciji znakov miotonije, značilni porazdelitvi mišične oslabelosti, dedovanju po dominantnem tipu in distrofičnih spremembah v mišicah. V otroštvu je potek bolezni lahko neugoden, pogosto ga spremlja duševna zaostalost. Do adolescence pride v ospredje mišična oslabelost. Pri funkcionalnih motnjah je indicirano zdravljenje z novokainom in kinidinom.

