Arnold-Chiari to wada wrodzona lub anomalia występująca podczas formowania się dziecka w łonie matki. Anomalia występuje z powodu ucisku mózgu, z tego powodu obszary czaszki są zdeformowane. Konsekwencje to: móżdżek i pień mózgu mocno się przesuń i wpadnij część potyliczna, wydajność jest pogorszona.
Wszystkie zabiegi należy wykonywać wyłącznie na zalecenie lekarza. Prawdziwy specjalista nie postawi trafna diagnoza bez odpowiedniego badania.
Skuteczne leczenie zespołu
Do chwili obecnej stosuje się dwa rodzaje leczenia: chirurgiczne, jeśli chodzi o, i zachowawcze.

Leczenie zachowawcze stosuje się, gdy choroba nie powoduje dużego dyskomfortu u pacjenta i nie wpływa na jego rozwój. Lekarz zaleca częstsze wychowanie fizyczne, ćwiczenia na koordynację mięśni. Przepisywane są również niektóre leki: przeciwbólowe, zwiotczające mięśnie, przeciwzapalne. Dodatkowo przepisywany jest kompleks, szczególnie grupa B, ponieważ odpowiadają one za procesy biochemiczne w organizmie i normalizują pracę ośrodkowego układu nerwowego.
Oczywiście takie wizyty nie pomogą całkowicie pozbyć się choroby, ale pozwolą ci obejść się bez interwencji chirurgicznej tak długo, jak to możliwe.
Jeśli zniekształcenie choroby postępuje, jest to pilne interwencja chirurgiczna. Zostanie wykonana operacja lub bajpas. Operacja pozwala rozwiązać dwa główne powody:
- Popraw wady, które przyczyniają się do ucisku czaszki i mózgu.
- Przywraca ruch płynu mózgowo-rdzeniowego do normalnego stanu.
Taka operacja jest dość powszechna, jej czas trwania nie przekracza dwóch godzin. Całkowity powrót do zdrowia następuje po kilku tygodniach. Dzięki operacji ciśnienie wewnątrzczaszkowe normalizuje się, zwiększa się przestrzeń w rdzeniu kręgowym i mózgu, choroba ustępuje.
Środki zapobiegawcze
Zawsze trzeba dbać o swoje zdrowie, a jeśli jest to okres, w którym kobieta nosi się pod sercem, to odpowiedzialność podwaja się. Istnieją pewne środki zapobiegawcze zapobiegające chorobie:
- włączaj do swojej diety więcej owoców i warzyw
- pić świeże soki, jeść nabiał i mięsa bogate w białko
- bierz witaminy prenatalne
- poddać się złe nawyki, Jeśli znajdują się jakiekolwiek
- pij tylko te leki, które są dozwolone w czasie ciąży i tylko zgodnie z zaleceniami lekarza
- przeprowadzić wszystkie niezbędne badania
Jeśli monitorujesz swoją dietę i prowadzisz pełnoprawny, zdrowy tryb życiażycia, zrób badania na czas i słuchaj lekarza, wtedy prawdopodobieństwo urodzenia zdrowego dziecka wzrasta wielokrotnie.
Tak więc zespół Arnolda-Chiari u płodu występuje przez rózne powody zarówno wrodzone, jak i nabyte. Choroba pierwszego i drugiego typu jest całkowicie uleczalna, jeśli zostanie wykonana niezbędna operacja. Aby zapobiec występowaniu patologii, przyszła mama musisz jak najbardziej dbać o swoje, dzięki czemu będzie to miało pozytywny wpływ na rozwój mózgu jej płodu.
25 lutego 2017 r Doktor Violetta
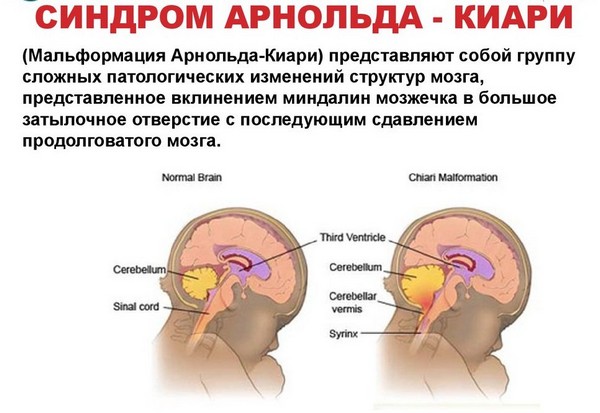
Malformacja Arnolda-Chiariego jest wadą rozwojową połączenia szyjno-rdzeniowego, charakteryzującą się przemieszczeniem migdałków móżdżku, a w niektórych przypadkach tułowia i komory IV poniżej poziomu otworu wielkiego.
J. Cleland jako pierwszy opisał wady rozwojowe w 1883 r. U 9 martwych niemowląt podczas sekcji zwłok stwierdził wydłużenie tułowia i zejście migdałków móżdżku do kanału kręgowego. Jednak twórczość J.Clelanda nie została dostrzeżona. Następnie w 1891 roku Hans von Chiari opisał wrodzoną anomalię polegającą na przepuklinie migdałków móżdżku poniżej poziomu otworu wielkiego.
Rodzaje wad rozwojowych
Malformacja Chiariego typu I- przemieszczenie migdałków móżdżku w dół przez otwór wielki do górnych odcinków rdzeń kręgowy. Tego typu wadom rozwojowym towarzyszy wodniak i zwykle ujawniają się one w okresie dojrzewania lub w wieku dorosłym. U nastolatków głównymi objawami są naruszenie zgięcia i zmniejszenie siły ramion, utrata bólu i wrażliwości na temperaturę w górnej połowie ciała i ramion. Dorośli zwykle skarżą się na ból szyi okolica potyliczna, nasilający się wraz z kaszlem, a także bólem rąk.
Malformacja Chiariego typu II charakteryzuje się przemieszczeniem robaka móżdżku, migdałków, czwartej komory i rdzeń przedłużony(części pnia mózgu) do otworu wielkiego. Ten typ, zwany także malformacją Arnolda-Chiariego, jest znacznie częściej związany z wodniakiem rdzenia niż z typem I i prawie zawsze wiąże się z przepukliną oponowo-rdzeniową. Przepuklina oponowo-rdzeniowa to wrodzone zaburzenie zamknięcia rdzenia kręgowego i kręgosłupa w okresie rozwoju płodowego. Objawy tej wady rozwojowej są oczywiste i zwykle pojawiają się zaraz po urodzeniu, wraz z krótkimi epizodami wstrzymania oddechu, osłabieniem odruchu gardłowego, mimowolnymi i szybkimi ruchami. gałki oczne w dół, zmniejszając siłę w ramionach.
Malformacja Chiariego typu III polega na przemieszczeniu móżdżku i części pnia mózgu wraz z oponami mózgowymi w oponie mózgowej, zlokalizowanej w okolicy szyjno-potylicznej.
Wcześniej zidentyfikowano wadę Chiariego typu IV, której towarzyszy niedorozwój móżdżku. Jednak obecnie większość autorów woli przypisywać ten typ wad rozwojowych anomalii Dandy-Walkera.
Malformacjom Arnolda-Chiariego typu II i III mogą towarzyszyć objawy dysplazji układu nerwowego: wielozakrętowość, heterotopia korowa, hipoplazja zwojów podstawy mózgu, dysgenezja ciała modzelowatego, patologia przegrody przezroczystej, pogrubienie połączenia międzywzgórzowego, pokrywa dziobowa (pokrywa krukowa). ), często stwierdza się obecność przegięcia akweduktu Sylwiusza (55%), torbiele otworu Magendiego, hipoplazję sierpa i niedrożność móżdżku, kręgów połowiczych, niskie położenie ogonowego rdzenia kręgowego na poziomie LIV– Kręgi V i poniżej.
Etiologia malformacji Chiariego
Etiologia choroby jest obecnie niejasna. Istnieją dowody na tę rolę czynnik genetyczny w etiologii tego zespołu. U trzech bliźniąt jednojajowych stwierdzono ektopię migdałków móżdżku do otworu wielkiego. Odkąd w 1883 roku po raz pierwszy opisano wadę rozwojową Clelanda, pojawiło się kilka teorii. Teoria, poparta badaniami Misao Nishikawy i współautorów, głosi, że z powodu dysplazji przyosiowej warstwy mezodermalnej lub pierwotnego uszkodzenia struktur odpowiedniego somitu powstaje nienormalnie mały tylny dół czaszki, struktury tyłomózgowia , wypełniając objętość tylnego dołu czaszki i kontynuując wzrost, schodzą do kanału potylicznego. Połączenie anomalii Chiariego typu II z oponowo-rdzeniowym wynika z faktu, że stopień dysplazji przyosiowej warstwy mezodermalnej w typie AK-II jest bardziej wyraźny niż w typie AK-I i stwierdza się nie tylko na poziomie tworzenia się kości potylicznej , ale także wzdłuż osi ciała na poziomie rzędów kręgów, co objawia się rozszczepem kręgosłupa, a także anomaliami szeregu innych struktury kostne I układ szkieletowy ogólnie.
Obraz kliniczny malformacji Chiariego
Objawy kliniczne AK – piszę pojawiają się najczęściej w okresie dojrzewania lub dorosłości. Objawy te pasują do takich zespołów neurologicznych, jak zespoły móżdżkowo-opuszkowe, nadciśnienie alkoholowe, jamisto-mieliczne, zespoły uszkodzenia nerwy czaszkowe. Zespół nadciśnieniowy płynu mózgowo-rdzeniowego objawia się bólem głowy, zwykle podpotylicznym i bólem szyi, nasilonym przez kaszel, kichanie i wysiłek, stojące dyski nerwy wzrokowe. Dysfunkcje pnia i nerwu czaszkowego objawiają się niestabilną oscylopsją, dysestezją nerwu trójdzielnego, utratą słuchu, szumem w uszach, zawrotami głowy, dysfagią, zatrzymaniem oddechu podczas snu, okresowymi omdleniami (często związanymi z kaszlem), zaburzeniami kontroli rytmu serca, ciśnieniem krwi podczas przejścia z pozycji poziomej do pozycja pionowa, zanik połowy języka, paraliż struny głosowe, stridor, spastyczny lub mieszany (bardziej w kończynach górnych) tetrapareza. Zaburzenia móżdżku- oczopląs, dyzartria, ataksja. Objawy towarzyszące torbieli syringomielicznej to drętwienie, zaburzenia czucia, zwykle typu dysocjacyjnego, a także neuroartropatia, dysfunkcja narządów miednicy, brak odruchów brzusznych i zanik mięśni. Jednocześnie wielu autorów zauważa rozbieżność między lokalizacją, długością torbieli, wskaźnikiem torbielowatym (stosunek przednio-tylnego rozmiaru torbieli do średnicy rdzenia kręgowego na poziomie torbieli ), z jednej strony, a strefa hipestezji, występowanie segmentowych zaburzeń powierzchownej wrażliwości, nasilenie hipotrofii mięśni i stopień niedowładu - z drugiej.
typu AK II objawia się u noworodków i wcześnie dzieciństwo objawy takie jak bezdech, stridor, obustronny niedowład strun głosowych, dysfagia neurogenna z zarzucaniem nosa, sinica podczas karmienia, oczopląs, niedociśnienie, osłabienie, spastyczność kończyn górnych, która może prowadzić do tetraplegii.
Wada rozwojowa Chiari III typ jest rzadki, jego objawy kliniczne są takie same jak w AK II.
Rozpoznanie malformacji Chiariego
Standard badanie rentgenowskie może ujawnić jedynie pośrednie oznaki malformacji AK, tomografia komputerowa nie daje również wyraźnej wizualizacji struktur tkanek miękkich. Powszechne wprowadzenie MRI do praktyki klinicznej umożliwiło rozwiązanie większości problemów związanych z diagnostyką anomalii Chiariego. Było to ułatwione dzięki dobrej wizualizacji struktur tylnego dołu czaszki, połączenia czaszkowo-kręgowego, rdzenia kręgowego i braku artefaktów ze struktur kostnych.
Leczenie malformacji Chiari
Możliwe jest jedynie leczenie malformacji Chiariego i związanej z nią jamistości rdzenia chirurgicznie. Operacja polega na odbarczeniu tylnego dołu czaszki lub założeniu zastawki do płynu mózgowo-rdzeniowego w przypadku współistniejącego wodogłowia.
Lokalna dekompresja wykonywana jest pod ogólne znieczulenie i polega na usunięciu części kości potylicznej, a także tylnych połówek I i/lub II kręgów szyjnych do miejsca, w którym zstępują migdałki móżdżku. Resekuje się również obniżone migdałki móżdżku, eliminując w ten sposób ucisk pnia mózgu. Ten efektywne działanie rozszerza otwór wielki i eliminuje ucisk pnia mózgu, rdzenia kręgowego i migdałków móżdżku. Operacja otwiera również oponę twardą, grubą błonę otaczającą mózg i rdzeń kręgowy. Do otwartej opony twardej wszywa się plaster z innej tkanki (sztucznej lub pobranej od samego pacjenta), aby płyn mózgowo-rdzeniowy mógł swobodniej przepływać.
Rzadziej wykonuje się operacje drenażowe płyn mózgowo-rdzeniowy od rozszerzonego rdzenia kręgowego do klatki piersiowej lub Jama brzuszna za pomocą specjalnego pustego bocznika z zastawką lub do przestrzeni dooponowej. Czasami te operacje są wykonywane etapami.
Według różnych autorów skuteczność dekompresji chirurgicznej waha się od 50 do 85%. Należy pamiętać, że leczenie chirurgiczne najlepiej przeprowadzić przed rozwojem poważnych zaburzeń neurologicznych, ponieważ później leczenie chirurgiczne przywrócenie funkcji jest niepełne lub w ogóle nie następuje, a głównym zadaniem operacji jest stabilizacja stanu neurologicznego pacjenta i zapobieganie dalszemu postępowi choroby.
Izraelskie Centrum Neurochirurgii i Neurologii Neuromed specjalizuje się w operacjach neurochirurgicznych anomalii Arnolda-Chiariego z wykorzystaniem innowacyjne metody w celu poprawy skuteczności i bezpieczeństwa leczenia.
Charakteryzuje się niedopasowaniem wielkości tylnego dołu czaszki do struktur mózgu znajdujących się w tym obszarze, w wyniku czego migdałki móżdżku i pnia mózgu schodzą do otworu wielkiego. Powoduje to ich naruszenie w tym zakresie. Często ta patologia w połączeniu z zaburzeniami rozwoju rdzenia kręgowego.
Większość pacjentów z zespołem Arnolda-Chiariego nie odczuwa żadnych objawów, a choroba jest najczęściej wykrywana przypadkowo, podczas diagnozowania innych chorób. Anomalia Chiari występuje rzadko: dotyka około 3-8 osób na sto tysięcy przypadków.
Jakie są przyczyny zespołu Arnolda-Chiariego?
Do chwili obecnej etiologia tej choroby nie została w pełni zbadana i wśród specjalistów nie ma zgody co do przyczyn choroby. Zakłada się, że anomalia Arnolda-Chiariego wynika z zaburzeń strukturalnych w mózgu i rdzeniu kręgowym. Rozwijają się podczas rozwoju płodu i są z nimi związane mutacje genetyczne lub niezbilansowana dieta kobiety w okresie ciąży, gdy w diecie brakuje składników odżywczych.
Według innej wersji anomalia Chiari występuje z powodu zwiększonego rozmiaru mózgu i niejako przemieszcza zawartość tylnego dołu czaszki przez otwór wielki.
Przejście drobnej wady do wyraźnej postać kliniczna może powodować wodogłowie. Jest to spowodowane zwiększeniem objętości mózgu w wyniku wzrostu komór. Ze względu na to, że z zespołem Chiari, oprócz dysplazji tkanka kostna połączenie czaszkowo-kręgowe jest słabo rozwinięte aparat więzadłowy w tym obszarze nawet najmniejsze urazowe uszkodzenie mózgu może pogorszyć penetrację migdałków móżdżku do otworu potylicznego.
Klasyfikacja anomalii Arnolda-Chiariego
Ta patologia jest podzielona na cztery typy:
- Anomalia Arnolda-Chiariego I stopnia- objawia się zejściem migdałków móżdżku poniżej poziomu dużego otworu potylicznego. Z reguły objawia się w okresie dojrzewania i później. Często towarzyszy wodniak, w którym płyn mózgowo-rdzeniowy gromadzi się w rdzeniu kręgowym, w jego kanale centralnym;
- Anomalia Arnolda-Chiariego II stopnia- objawia się niemal natychmiast po urodzeniu dziecka, w pierwszych dniach jego życia. W tej postaci choroby rdzeń przedłużony, robak móżdżku i komora IV wychodzą przez otwór wielki oprócz migdałków. Ten typ choroby często wiąże się z wodniakiem rdzenia i prawie zawsze wiąże się z obecnością wrodzonej przepukliny kręgosłupa;
- anomalia Arnolda-Chiariego III stopnia- Objawia się tym, że móżdżek i rdzeń przedłużony, schodzący poniżej otworu wielkiego, znajdują się w oponie mózgowej okolicy szyjno-potylicznej;
- anomalia Arnolda-Chiariego IV stopnia- objawia się niedorozwojem (hipoplazją) móżdżku, w którym nie ma przemieszczenia w kierunku ogonowym. Niektórzy eksperci przypisują tę wadę zespołowi Dandy'ego-Walkera, gdy oprócz hipoplazji móżdżku występują wrodzone torbiele tylnego dołu czaszki i hipercefalia.
Anomalie Chiari I i II stopnia są dość często łączone różne patologie układ nerwowy, np.:
- polimikrozakręt;
- heterotopia kory mózgowej;
- defekty ciała modzelowatego;
- cysty otworu Mogendiego;
- hipoplazja struktur podkorowych;
- załamanie akweduktu Sylwiańskiego itp.
Anomalia Arnolda-Chiariego: objawy
Objawy anomalii Arnolda-Chiariego różnią się w zależności od rodzaju choroby. W praktyka kliniczna najczęstsza anomalia Chiariego typu I, w której zajęte są nerwy czaszkowe, obserwuje się także zespoły nadciśnienia jamisto-mielicznego, móżdżkowo-opuszkowego i mózgowo-rdzeniowego. Tego typu chorobę najczęściej wykrywa się już w wieku dorosłym lub w okresie dojrzewania.
Zespół nadciśnienia alkoholowego towarzyszący anomalii Chiari I charakteryzuje się bólem głowy w okolicy potylicznej i szyjnej. Ból nasila się podczas napięcia mięśni szyi, na przykład podczas kaszlu lub kichania. Często występują wymioty, które nie mają nic wspólnego ze spożyciem pokarmu i jego charakterem. Podczas badania pacjent ma zwiększone napięcie mięśni szyjnych. Zaburzenia móżdżkowe objawiająca się dyzartrią (niewyraźna mowa), mimowolne ruchy i zaburzenia motoryczne.
Uszkodzenie pnia mózgu oraz znajdujących się w nim korzeni i jąder nerwów czaszkowych powoduje zaburzenia widzenia, niespójną mowę, zaburzenia połykania, utratę słuchu, ogólnoustrojowe zawroty głowy z uczuciem obracających się obiektów, szumy uszne, zespół bezdechu sennego, powtarzające się krótkoterminowe straty przytomność, zapaść ortostatyczna. Pacjenci zgłaszają nasilenie szumów usznych i zawrotów głowy podczas obracania głowy, co może nawet powodować omdlenia. Możliwe są niedowłady krtani i trudności w oddychaniu.
Anomalie Arnolda-Chiari II i III stopnia są podobne w swoim działaniu objawy kliniczne. Stają się zauważalne już w pierwszych dniach życia dziecka. Anomalia Chiari II charakteryzuje się głośnym oddychaniem, okresami krótkiego zatrzymania oddechu, niedowładem neuropatycznym krtani, problemami z połykaniem podczas wrzucania do nosa płynnego pokarmu. Noworodki mają oczopląs, zwiększony napięcie mięśniowe, sinica skóry podczas karmienia. Może być również obecny zaburzenia ruchu różnym stopniu.
Anomalia Chiari III jest cięższą postacią choroby i często rozwój płodu nie jest zgodny z życiem.
Jak diagnozuje się zespół Arnolda-Chiariego?
Diagnozując tę patologię, badania lekarskie ani zestaw (REG, EEG, Echo-EG) nie dostarczają danych, na podstawie których można postawić trafną diagnozę. Pozwalają zidentyfikować jedynie oznaki wodogłowia.
Badanie rentgenowskie pozwala wykryć jedynie ubytki kostne towarzyszące anomalii Chiariego. Przed wprowadzeniem tomografii w praktyka neurologiczna diagnostyka tę chorobę było trudne. Teraz możliwa jest dokładna diagnoza.
Zespół Arnolda-Chiariego: leczenie
Jak wspomniano powyżej, choroba w większości przypadków przebiega bezobjawowo, dlatego bardzo często zespół Arnolda-Chiariego nie wymaga leczenia. W przypadku bólu w okolicy szyjnej i potylicznej przeprowadza się leczenie zachowawcze. Obejmuje leki przeciwbólowe, zwiotczające mięśnie i leki przeciwzapalne.
Schorzenia neurologiczne (zaburzenia napięcia i wrażliwości mięśni, niedowłady itp.), a także w przypadku nieskutecznego leczenia zespołów bólowych metoda konserwatywna stosowana jest interwencja chirurgiczna, której celem jest zatrzymanie procesu zmiany strukturalne w kręgosłupie i mózgu oraz w celu ustabilizowania objawów. Po pomyślnym wyniku operacji zmniejsza się nacisk na móżdżek i przywracany jest odpływ płynu mózgowo-rdzeniowego.
Istnieją choroby związane z nieprawidłowym ułożeniem narządów, co powoduje dysfunkcję w tych obszarach. Jedną z takich dolegliwości jest zespół Arnolda Chiari (wada Chiariego). Charakteryzuje się nieprawidłowym położeniem móżdżku i pnia mózgu w czaszce, przez co wystają one nieznacznie do otworu wielkiego. Zjawisku temu towarzyszą ataki bólu w tylnej części głowy, niespójna mowa, zaburzenia koordynacji, osłabienie mięśni krtani i inne objawy. Choroba jest podzielona na 4 odmiany, z których każda ma swoje własne objawy i leczenie.
Anomalię Chiari diagnozuje się po prostu i w tym celu wystarczy wykonać MRI. Patologię można wyleczyć jedynie za pomocą interwencji chirurgicznej.
Istnieje pewna różnica pomiędzy rdzeniem kręgowym a mózgiem, czyli otworem wielkim. W tym miejscu łączą się, a nad nim zlokalizowany jest tylny dół czaszki, zawierający móżdżek, mostek i rdzeń przedłużony. W przypadku choroby Chiari tkanka mózgowa wpada do otworu wielkiego, który uciska rdzeń przedłużony i rdzeń kręgowy zlokalizowane w tym obszarze. Często z powodu tego procesu pogarsza się odpływ płynu mózgowo-rdzeniowego z głowy (płynu mózgowo-rdzeniowego), co prowadzi do rozwoju wodogłowia (wodogłowie).
Zespół Chiari odnosi się do wrodzonych patologii połączenia czaszki i wyższa liga kręgosłup.
Diagnozę taką stawia się dość rzadko i według statystyk stwierdza się ją u 10 noworodków na 120 tys. osób.
Możliwe jest stwierdzenie obecności choroby w ciągu jednego lub dwóch dni od momentu urodzenia, ale niektóre typy choroby występują dopiero w wieku dorosłym. Najczęściej choroba Arnolda Chiari rozwija się wraz z jamistością rdzenia, która charakteryzuje się pustymi obszarami w istocie rdzenia kręgowego i rdzenia przedłużonego.
Powody rozwoju
Anomalia Arnolda Chiari nie została jeszcze szczegółowo zbadana i nikt nie jest w stanie podać dokładnych czynników wpływających na rozwój choroby. Istnieje wiele założeń, na przykład niektórzy eksperci uważają, że patologia jest spowodowana zbyt małym rozmiarem tylnego dołu czaszki. W rezultacie tkanki nie mają wystarczającej ilości miejsca i schodzą do otworu wielkiego. Alternatywna teoria głosi, że mózg od urodzenia jest większy niż normalnie. Dlatego wpycha część swoich tkanek do otworu wielkiego.
Ze względu na stagnację płynu mózgowo-rdzeniowego i późniejsze powstawanie wodogłowia patologia ma bardziej wyraźne objawy. Wszakże z powodu tej choroby komory zwiększają się, a co za tym idzie, rozmiar mózgu. Proces ten wzmaga wyciskanie migdałków móżdżku do otworu wielkiego. Urazy głowy mogą również pogorszyć przebieg choroby. Choroba Chiari charakteryzuje się nie tylko nieprawidłowym rozwojem tkanek przejścia między rdzeniem kręgowym a mózgiem, ale także anomaliami aparatu więzadłowego. Wszelkie uszkodzenia w okolicy głowy jeszcze bardziej dociskają móżdżek do okolicy potylicznej, przez co choroba objawia się silniej.
Odmiany procesu patologicznego
Wada rozwojowa Chiari ma następujące rodzaje rozwoju:
- Pierwszy widok. W przypadku anomalii Arnolda Chiari typu 1 migdałki móżdżku często wchodzą do otworu wielkiego. Choroba objawia się głównie w okresie dojrzewania, a czasami już w wieku dorosłym. Niezwykle często w przypadku malformacji Arnolda Chiariego typu 1 występuje wodniak;
- Drugi rodzaj. Diagnozę tę stawia się już w pierwszych dniach od chwili narodzin dziecka. Anomalia Arnolda Chiariego typu 2 jest znacznie poważniejsza niż typ 1. W jej przypadku do jamy potylicznej wpada również fragment móżdżku (oprócz migdałka), a także komora czwarta i rdzeń przedłużony. Wodogłowie w tego typu chorobach występuje znacznie częściej. Przyczyną tego zjawiska w większości przypadków jest wrodzona;
- Trzeci rodzaj. Różni się od drugiego typu tym, że tkanki, które opadły do otworu potylicznego, wchodzą do opon mózgowo-rdzeniowych (przepukliny), która znajduje się w części szyjno-potylicznej;
- Widok czwarty. Jego istotą jest słabo rozwinięty móżdżek, który w przeciwieństwie do innych typów chorób nie przemieszcza się do otworu wielkiego. Niektórzy eksperci uważają, że choroba Chiari typu 4 jest częścią zespołu Dandy-Walkera. Charakteryzuje się rozwojem cyst zlokalizowanych w okolicy tylnego dołu czaszki i obrzękiem mózgu.
Drugi i trzeci typ anomalii często występują w połączeniu z zły rozwój inne tkanki układu nerwowego, a mianowicie:
- Nietypowe położenie tkanek kory mózgowej;
- Patologie ciała modzelowatego;
- Polimikrogyria (wiele małych zwojów);
- Niedorozwój tkanek struktur podkorowych, a także sierp móżdżku.
Objawy
Najczęściej diagnozowana jest anomalia Arnold Chiari 1 stopnia. Łączy w sobie następujące syndromy:
- Syringomyeliczny;
- nadciśnienie CSF;
- Móżdżkowo-opuszkowy.
Na tym tle czaszkowo-mózgowy włókna nerwowe i żyć bez objawów tej patologii aż do okresu dojrzewania. U niektórych osób pierwsze oznaki stają się widoczne po 20 latach.
Zespół nadciśnienia alkoholowego, charakterystyczny dla anomalii Arnolda Chiari, objawia się takimi objawami:
- Ból w okolicy szyjnej i potylicznej, który objawia się podczas kaszlu i kichania, a także na skutek napięcia mięśni w tym miejscu;
- bezprzyczynowe wymioty;
- napięcie mięśni szyi;
- niespójna mowa;
- Zaburzona koordynacja ruchów;
- Niekontrolowany ruch gałek ocznych (oczopląs).
Z biegiem czasu, z powodu uszkodzenia pnia mózgu i otaczających nerwów, osoba doświadcza następujących objawów:
- niedowidzenie;
- Rozwidlony obraz przed oczami (diplopia);
- problemy z połykaniem;
- Utrata słuchu;
- Częste zawroty głowy;
- Hałas w uszach;
- zaburzenia snu połączone z zatrzymaniem oddechu przez nos i usta na 10 lub więcej sekund;
- Utrata przytomności;
- Niewystarczający dopływ krwi do mózgu, objawiający się niskim poziomem ciśnienie krwi podczas zmiany pozycji ciała.
W przypadku anomalii Arnolda Chiari objawy nasilają się z powodu ostrych skrętów głowy, a choroba objawia się w następujący sposób:
- Zwiększone zawroty głowy;
- Hałas w uszach staje się głośniejszy;
- Często traci przytomność;
- Połowa języka ulega zmianom zanikowym (zmniejszeniu rozmiaru);
- Mięśnie krtani słabną, co utrudnia oddychanie, a głos jest ochrypły;
- Osłabienie mięśni kończyn, głównie górnych.
Często patologia charakteryzuje się zespołem syringomielicznym, w tym przypadku objawia się następująco:
- Pogorszenie wrażliwości;
- Niedobór białka i energii (hipotrofia) tkanki mięśniowej;
- Drętwienie;
- Osłabienie lub całkowity brak odruchów brzusznych;
- Zaburzenia miednicy;
- Artropatia neurogenna (deformacja stawów).
Zespół móżdżkowo-opuszkowy występujący w chorobie Chiari objawia się następującymi objawami:
- Naruszenie funkcji nerwu trójdzielnego;
- Nerwowość przedsionkowo-ślimakowa;
- problemy z koordynacją;
- oczopląs;
- Zawroty głowy.
Drugi i trzeci stopień choroby objawia się podobnie, jednak pierwsze objawy pojawiają się już po 2-3 dniach od urodzenia. W przypadku anomalii typu 2 Arnold Chiari ma swoje własne cechy wyróżniające:
- Głośny oddech, który czasami zatrzymuje się na 10-15 sekund;
- Obustronne osłabienie mięśni krtani, w wyniku czego pojawiają się problemy z połykaniem, a płynny pokarm często wrzucany jest do nosa;
- oczopląs;
- Utwardzanie mięśni górne kończyny ze względu na zwiększony ton;
- zmienia kolor na niebieski skóra(sinica);
- Trudności w poruszaniu się aż do paraliżu większości ciała (tetraplegia).
Trzeci rodzaj anomalii jest znacznie poważniejszy i rzadko zgodny z życiem z powodu poważnych naruszeń.
Diagnostyka
W dawnych czasach niezwykle trudno było zdiagnozować patologię, ponieważ ankieta, badanie i standardowe metody badań nie dawały wielu wyników. Pokazali obecność wysokie ciśnienie krwi i obrzęk mózgu. Rentgen nieco uprościł zadanie, ponieważ wykazał deformacje kości w czaszce, ale to nie pozwoliło nam być całkowicie pewnymi diagnozy. Sytuacja uległa zmianie wraz z pojawieniem się badań topograficznych. Przecież taka metoda diagnostyczna umożliwiła pełne zbadanie formacji w tylnym dole czaszki. Dlatego MRI (rezonans magnetyczny) jest uważany za niezbędną metodę badawczą różnicującą zespół Chiariego od innych procesów patologicznych.
Za pomocą rezonansu magnetycznego będziesz musiał zbadać obszar szyi i klatki piersiowej. Przecież w tych miejscach kręgosłupa często występują meningocele, a także torbiele syringomieliczne. Podczas badania lekarz musi nie tylko zweryfikować obecność zespołu Chiari, ale także wykluczyć inne procesy patologiczne które często są z tym związane.
Przebieg terapii
W przypadku anomalii Arnolda Chiari leczenie nie jest wymagane tylko wtedy, gdy patologia przebiega bezobjawowo. W takiej sytuacji należy unikać uderzeń i urazów głowy i szyi, aby nie pogorszyć przebiegu choroby.
Jeśli choroba przebiega z minimalnymi objawami, a mianowicie z łagodnym bólem, konieczne jest zastosowanie kuracji lekami o działaniu przeciwbólowym i przeciwzapalnym. Nie przeszkadzaj w wyznaczaniu środków zwiotczających mięśnie w celu rozluźnienia mięśni.
W ciężkie przypadki gdy choroba postępuje z wyraźnymi objawami (osłabienie mięśni, dysfunkcja nerwów czaszkowych itp.), konieczna będzie interwencja chirurgiczna. Podczas operacji lekarz rozszerzy otwór wielki poprzez usunięcie fragmentu kości potylicznej. Jeśli chcesz usunąć nacisk z pnia mózgu i rdzenia kręgowego, musisz usunąć część migdałków móżdżku i przednią połowę 2 górnych kręgów. Aby znormalizować krążenie płynu mózgowo-rdzeniowego, chirurg będzie musiał wykonać łatkę w oponie twardej.
W niektórych przypadkach leczenie chirurgiczne przeprowadza się za pomocą operacji bajpasów. Taka operacja jest odgałęzieniem płynu mózgowo-rdzeniowego za pomocą drenażu, w wyniku czego przestaje on stagnować.
Prognoza

Wiele osób cierpiących na zespół Arnolda Chiari zastanawia się, ile im jeszcze życia pozostało. Na to pytanie można odpowiedzieć w zależności od rodzaju patologii i ciężkości przebiegu. Ważnym czynnikiem jest także terminowość interwencji chirurgicznej.
Osoby cierpiące na chorobę Chiari typu 1 często mają średnią długość życia, ponieważ patologia może przebiegać bezobjawowo. Jeśli występuje choroba typu 1 lub 2 objawy neurologiczne ważne jest, aby operację wykonać jak najszybciej. W końcu powikłania związane z mózgiem i rdzeniem kręgowym są praktycznie nieuleczalne. Trzeci rodzaj choroby najczęściej kończy się śmiercią pacjenta przy urodzeniu.
Choroba Arnolda Chiariego jest wadą wrodzoną, którą należy leczyć już w momencie pojawienia się pierwszych objawów. W przeciwnym razie możesz na zawsze pozostać niepełnosprawny lub stracić życie.
Anomalia Arnolda-Chiariego jest wrodzonym zaburzeniem rozwoju romboidalny mózg objawiający się niedopasowaniem wielkości tylnego dołu czaszki do struktur mózgu znajdujących się w tym obszarze, co prowadzi do opadania pnia mózgu i migdałków móżdżku do otworu wielkiego i ich naruszenia na tym poziomie.
W większości przypadków wada łączy się z wodogłowiem i nieprawidłowościami w rozwoju rdzenia kręgowego. Przyczyną może być wrodzona dysplazja (naruszenie) szerokiego otworu potylicznego, którego rozmiar staje się znacznie większy niż normalnie.
Po raz pierwszy został opisany przez N. Chiari w 1896 roku. Schorzenie to charakteryzuje się przemieszczeniem doogonowym rdzenia przedłużonego, mostu i robaka móżdżku, gdy wszystkie te struktury znajdują się w odcinku szyjnym kręgosłupa.
Częstość występowania tej choroby wynosi od 3,3 do 8,2 obserwacji na 100 000 mieszkańców.
Nie ustalono rzeczywistej częstości występowania poszczególnych typów zespołu Arnolda-Chiariego i ogólnie częstości występowania tej wady. Jedną z przyczyn braku takich danych jest odmienne podejście do klasyfikacji tej wady. Według Klasyfikacja międzynarodowa chorób, zespół Arnolda-Chiariego ma odrębny kod (Q07.0), ale jest w nim zdefiniowany jako „... stan patologiczny, przy którym następuje wzrost ciśnienie śródczaszkowe w wyniku guza wewnątrzczaszkowego, okluzyjnych postaci wodogłowia, procesu zapalnego, który w niektórych przypadkach prowadzi do zaklinowania móżdżku i rdzenia przedłużonego w otworze wielkim. W ultrasonograficznej literaturze prenatalnej nie udało się dotychczas znaleźć opisów przypadków rozpoznania prenatalnego zespołu Arnolda-Chiariego, które w pełni odpowiadałyby tym cechom.
Cechy morfologiczne różnych typów wady Arolda-Chiariego determinują możliwości wykrywania prenatalnego i rokowanie na całe życie.
Przyczyny zespołu Arnolda-Chiariego nie zostały w pełni ustalone. Z reguły nie można wykryć nieprawidłowości chromosomalnych w tej patologii.
Patogeneza(co się dzieje?) podczas Anomalii Arnolda-Chiari:
Do chwili obecnej patogeneza patologii nie została ostatecznie ustalona. Najprawdopodobniej te czynniki patogenetyczne trzy:
pierwsza to dziedziczne wrodzone osteoneuropatie,
drugi - urazowe urazy części klinowo-sitowej i klinowo-potylicznej stoczni na skutek urazu porodowego,
trzeci to hydrodynamiczny wpływ płynu mózgowo-rdzeniowego na ściany kanału centralnego rdzenia kręgowego.
Cechy anatomiczne anomalii Chiariego
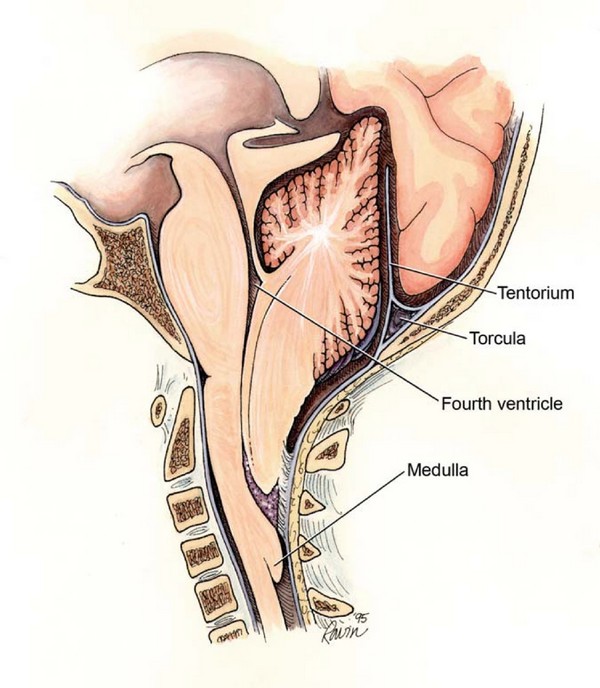
Móżdżek położony jest w tylnym dole czaszki. (ŚŚ)
Migdałki to dolna część móżdżku. Zwykle znajdują się nad dużym otworem potylicznym. W anomalii Chiari migdałki móżdżku znajdują się poniżej otworu wielkiego, w kanale kręgowym.
Otwór wielki stanowi swego rodzaju granicę pomiędzy czaszką a kręgosłupem, pomiędzy mózgiem a rdzeniem kręgowym. Nad otworem wielkim znajduje się tylny dół czaszki, poniżej kanału kręgowego.
Na poziomie otworu wielkiego dolna część pnia mózgu (rdzeń przedłużony) przechodzi do rdzenia kręgowego. Zwykle płyn mózgowo-rdzeniowy (CSF) krąży swobodnie w przestrzeniach podpajęczynówkowych mózgu i rdzenia kręgowego. Na poziomie otworu wielkiego łączą się przestrzenie podpajęczynówkowe mózgu i rdzenia kręgowego, co zapewnia swobodny odpływ płynu mózgowo-rdzeniowego z mózgu.
W anomalii Chiari nisko położone migdałki móżdżku utrudniają swobodny przepływ płynu mózgowo-rdzeniowego pomiędzy mózgiem a rdzeniem kręgowym. Migdałki blokują otwór wielki, podobnie jak korek zamyka szyjkę butelki. W rezultacie odpływ płynu mózgowo-rdzeniowego zostaje zakłócony i rozwija się wodogłowie.
Objawy anomalii Arnolda-Chiariego:
Chiari (Chiari) zidentyfikowała cztery typy anomalii wraz ze szczegółowym ich przedstawieniem. Lekarze do dziś posługują się tą klasyfikacją.
1. Anomalia Arnolda-Chiariego typu I polega na zejściu struktur PCF do kanału kręgowego poniżej płaszczyzny otworu wielkiego.
2. W przypadku anomalii Arnolda-Chiari typu II - dochodzi do ogonowego przemieszczenia dolnych części robaka, rdzenia przedłużonego i komory IV, często rozwija się wodogłowie.
3. Anomalia Arnolda-Chiariego typu III jest rzadka i charakteryzuje się dużym przemieszczeniem doogonowym wszystkich struktur tylnego dołu czaszki.
4. Anomalia Arnolda-Chiariego typu IV - hipoplazja móżdżku bez przemieszczenia w dół.
Anomalie typu III i IV są zwykle nie do pogodzenia z życiem.
U około 80% pacjentów anomalia Arnolda-Chiariego łączy się z patologią rdzenia kręgowego - jamistością rdzenia, która charakteryzuje się tworzeniem cyst w rdzeniu kręgowym, powodując postępującą mielopatię. Torbiele te powstają, gdy struktury tylnego dołu czaszki opadają i ulegają uciskowi szyjny rdzeń kręgowy.
Typowy obraz kliniczny Anomalia Arnolda-Chiariego charakteryzuje się następującymi cechami objawy:
Ból szyi nasilający się przy kaszlu, kichaniu,
Zmniejszenie wrażliwości bólowej i temperaturowej w kończynach górnych,
spadek siła mięśni w kończynach górnych
Spastyczność kończyn górnych i dolnych,
omdlenia, zawroty głowy,
Zmniejszona ostrość wzroku
W bardziej zaawansowanych przypadkach dołączają się: epizody bezdechu (krótkie zatrzymanie oddechu), osłabienie odruchu gardłowego, mimowolne, szybkie ruchy gałek ocznych.
Możliwe konsekwencje, powikłania:
1. Na tle nasilających się objawów nadciśnienia wewnątrzczaszkowego (czasami bez niego), postępującej dysfunkcji móżdżku i ucisku rdzenia kręgowego w odcinku szyjnym obserwuje się porażenie nerwów czaszkowych.
2. Czasami anomalia Arnolda-Chiari łączy się z wadami kości - potylicą atlasu i wyciskiem podstawnym (lejkowate zagłębienie stawu łokciowego i stawu czaszkowo-rdzeniowego).
3. Anomalie kręgosłupa, deformacje stóp.
Diagnoza anomalii Arnolda-Chiariego :
Czasami anomalia Chiari nie objawia się w żaden sposób i jest wykrywana przypadkowo podczas procedur diagnostycznych.
Obecnie metodą z wyboru w diagnostyce tej patologii jest MRI mózgu odcinka szyjnego i piersiowy rdzeń kręgowy (aby wykluczyć jamistość rdzenia).
Leczenie anomalii Arnolda-Chiariego :
Jeśli jedynym objawem choroby jest niewielka intensywność zespół bólowy, w leczeniu stosuje się terapię zachowawczą, która obejmuje różne schematy przy stosowaniu niesteroidowych leków przeciwzapalnych i leków zwiotczających mięśnie.
Jeżeli w ciągu 2-3 miesięcy nie ma efektu leczenia zachowawczego lub u pacjenta występują deficyty neurologiczne (drętwienie, osłabienie kończyn itp.), wskazana jest operacja.
Celem operacji jest– laminektomia, kraniektomia dekompresyjna tylnego dołu czaszki i chirurgia plastyczna dysku twardego opony mózgowe. Przy takiej operacji zwiększa się objętość tylnego dołu czaszki i rozszerzenie otworu wielkiego, w wyniku czego ustanie ucisk struktur nerwowych i normalizacja przepływu płynu mózgowo-rdzeniowego. W przypadku współistniejącego wodogłowia wykonuje się operację bajpasów.
W Izraelu pacjentom oferuje się oszczędne i wysokiej jakości leczenie, które po leczeniu umożliwia pacjentom pełne życie. Leczenie chirurgiczne zespołu Arnolda-Chiariego przeprowadza się za pomocą endoskopu, minimalizując traumatyczny efekt leczenia operacyjnego. Metoda małoinwazyjnego leczenia chirurgicznego prowadzona w izraelskich klinikach pozwala pacjentom z anomalią Arnolda-Chiariego na prowadzenie w przyszłości pełnowartościowego życia, nawet bez wsparcia lekowego.
Symptomatologia zespołu Arnolda-Chiariego
Wrodzone anomalie kości i stawów są klinicznie mniej oczywiste same w sobie, a znacznie bardziej z ich powodu poważne powikłania w centralnym i peryferyjnym system nerwowy. Objawy neurologiczne są najsurowiej tolerowane przez pacjentów i determinują niekorzystny przebieg tego zespołu. Ogólnie rzecz biorąc, początek choroby jest powolny i nietypowy.
Symptomatologia, początkowo bardzo niewyraźna, a nawet nieobecna przez długi czas, jest często wykrywany w wyniku działania szeregu czynników rozstrzygających, takich jak urazowe uszkodzenie mózgu lub infekcje nosogardzieli.
Pierwsza manifestacja, która zwraca uwagę na obecność wrodzona anomalia bezpośrednio po porodzie jest obecność przepukliny oponowo-rdzeniowych (przepukliny kręgosłupa). Później odnotowuje się inne zjawiska kliniczne wskazujące na obecność anomalii kostnych i neurologicznych, a mianowicie:
- rozszczep kręgosłupa,
- boczne pochylenie głowy
- skrzywienie gałek ocznych,
- ból głowy oraz przerywany lub przemijający ból szyi (szczególnie u starszych dzieci i dorosłych), który pojawia się wraz z ruchami głowy;
- nudności wymioty.
U wielu pacjentów w związku z blokadą krążenia płynu mózgowo-rdzeniowego pomiędzy komorą IV a zbiornikami podstawy czaszki już w pierwszych miesiącach życia dochodzi do mimowolnego wodogłowie wewnętrzne co powoduje pojawienie się bardzo wielu różnorodnych zjawisk neurologicznych. Stopniowo czaszka dziecka powiększa się i pojawiają się trudności w karmieniu, a także problemy z oddychaniem, a opona mózgowa (jeśli występuje) może ulec owrzodzeniu.
Objawy nadciśnienia wewnątrzczaszkowego:
- silne bóle głowy,
- przekrwienie brodawek lub zanik nerwu wzrokowego (późny),
- towarzyszy postępujące upośledzenie funkcja wizualna aż do całkowitej ślepoty.
Objawy móżdżkowe:
- zawroty głowy;
- ataksja podczas chodzenia i w pozycji ortostatycznej;
- dyzartria;
- zaburzenia połykania,
- celowe drżenie,
- oczopląs.
Objawy w obwodowym układzie nerwowym:
- parestezje, znieczulenie, niedowład lub porażenie typu spastycznego,
- wzmożone odruchy ścięgniste kości,
- obecność odruchu Babińskiego.
Manifestacje w zakresie nerwów czaszkowych:
- jednostronny lub rzadziej obustronny paraliż twarzy;
- paraliż nerwy okoruchowe, wyrażany najczęściej przez zez wewnętrzny lub podwójne widzenie.
Rozpoznanie zespołu Arnolda-Chiariego.
Nakłucie lędźwiowe oraz badania biochemiczne i bakteriologiczne płynu mózgowo-rdzeniowego w większości przypadków nie dostarczają istotnych danych. Poza tym aplikacja nakłucie lędźwiowe może pogorszyć stan pacjenta, a nawet spowodować śmierć w wyniku nagłego spadku ciśnienia i całkowitego wniknięcia migdałków móżdżku i rdzenia przedłużonego do kanału kręgowego.
Zwykłe badanie rentgenowskie ujawnia następujące aspekty: mały tylny dół czaszki; poszerzenie otworu wielkiego i kanału kręgowego; wodogłowie (duża czaszka z rozbieżnymi szwami); odciski palców na płytce kostnej czaszki; spłaszczenie siodła tureckiego; rozszczep kręgosłupa szyjnego, grzbietowego i lędźwiowego.
Dodatkowe badania RTG (mieloencefalografia gazowa) są przeciwwskazane u dzieci poniżej 2. roku życia. Natomiast u starszych dzieci i dorosłych określają pośrednie i bezpośrednie objawy wodogłowia, przemieszczenie rdzenia przedłużonego i migdałków móżdżku, a także ucisk rdzenia kręgowego w odcinku szyjnym.
Badanie patologiczne. Z anatomicznego i topograficznego punktu widzenia zespół Arnolda-Chiariego występuje w czterech dobrze zindywidualizowanych typach, a mianowicie:
- Pierwszy typ, w którym dochodzi do rozciągnięcia i pominięcia migdałków móżdżku bez przemieszczenia rdzenia przedłużonego. Ten typ występuje najczęściej u starszych dzieci i dorosłych. Z punkt kliniczny wzroku, może pozostać bezobjawowa przez całe życie, a jej wykrycie może nastąpić całkiem przypadkowo.
- Drugi typ, w którym dolna część móżdżku i rdzeń przedłużony przemieszczają się przez duży otwór potyliczny do kanału kręgowego. Ten typ występuje częściej u niemowląt i klinicznie objawia się wodogłowiem, a często także obecnością przepukliny oponowo-rdzeniowych.
- Trzeci typ, z całkowitą penetracją móżdżku do przepukliny oponowo-rdzeniowych kręgów szyjnych.
- Czwarty typ, w którym obserwuje się hipoplazję móżdżku, spowodowaną jego całkowitą przepukliną; nie można rozróżnić robaka móżdżku, a migdałki i płat móżdżku są ledwo widoczne. Ta forma jest niezwykle rzadka.
Oprócz anomalii kości potylicznej i kręgów wpływających na móżdżek i rdzeń przedłużony, istnieją inne anomalie czaszkowo-mózgowe, a mianowicie lutowanie pierwszego kręgu z kością potyliczną, ruch kręgosłupa w górę do obszaru czaszki z powodu hipoplazji kości potylicznej kości potylicznej; zespolenie dwóch lub trzech kręgów (najczęściej 2. i 3. kręgów szyjnych), jak ma to miejsce w zespole Klippela-Feila; rozszczep kręgosłupa szyjnego, grzbietowego lub lędźwiowego.
Kurs i prognoza Zespół Arnolda-Chiariego . Przebieg choroby jest powolny. Pojawienie się zespołu u noworodka uniemożliwia jego przeżycie, a jego przebieg szybko prowadzi do śmierci.
W przypadku wolniejszego przebiegu zespół Arnolda-Chiariego jest powikłany pojawieniem się przewlekłego zapalenia pajęczynówki i zmian miąższowych aksonu, co nabiera szczególnego znaczenia, gdy w organizmie występują zaburzenia krążenia tkanka nerwowa. Zwykle w takich przypadkach zaburzenia neuropsychiatryczne są wykrywane późno i objawiają się paraplegią, tetraplegią i upośledzeniem umysłowym.
Leczenie Zespół Arnolda-Chiariego . jedyny leczenie afektywne jest zabiegiem chirurgicznym. Wskazania do operacji nie są stawiane na podstawie rentgenowskiego określenia anomalii kostnych, ale tylko wtedy, gdy tym ostatnim towarzyszą ciężkie objawy neurologiczne. Interwencja chirurgiczna polega na kraniotomii potylicznej połączonej z wysoką laminektomią; twardą skorupę wycina się i pozostawia otwartą. Zrosty włókniste, często rozległe, występujące wokół otworu wielkiego, krzyżują się i złuszczają.
W obecności ciężkie wodogłowie komorę wyprowadza się za pomocą zastawki Holtera lub Pudenza (wg metoda klasyczna operacja wodogłowia).
Choć w niektórych przypadkach natychmiastowe rezultaty są korzystne, to jednak każda interwencja chirurgiczna w przypadku zespołu Arnolda-Chiariego wiąże się z dużym ryzykiem pooperacyjnym, ze względu na zaburzenia w rdzeniu przedłużonym, które mogą wystąpić w najbliższym czasie. okres pooperacyjny i często powodują śmierć.

