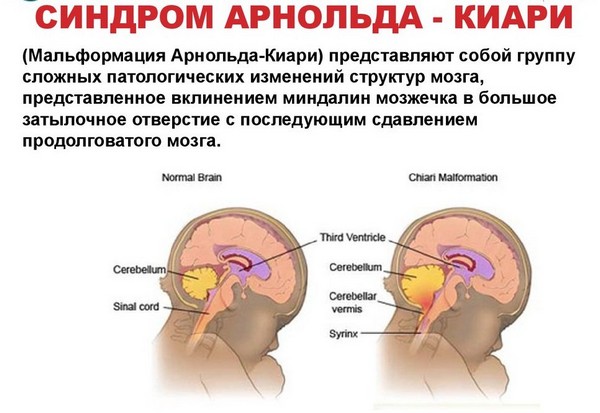
Арнольд-Киари – это врожденная или аномалия, которая происходит еще в период формирования ребенка внутри утробы матери. Аномалия происходит по причине сдавливания головного мозга, из-за этого деформируются черепные отделы. Последствия таковы: мозжечок и мозговой ствол сильно смещаются и опускаются в затылочную часть, нарушается работоспособность .
Все процедуры следует проводить четко по рекомендации врача. Настоящий специалист не будет ставить точный диагноз, не проведя должного обследования.
Эффективное лечение синдрома
На сегодняшний момент используют два вида лечения: хирургическое, когда дело доходит до , и консервативное.

Консервативное лечение используют в том случае, когда болезнь не причиняет больному сильного дискомфорта и не отражается на его развитии. Врач рекомендует чаще заниматься физкультурой, упражнениями для мышечной координации. Также прописываются некоторые препараты: обезболивающее, для расслабления мышц, противовоспалительное средство. Дополнительно назначается комплекс , особенно группы B, так как они отвечают в организме за биохимические процессы и нормализуют работу центральной нервной системы.
Конечно, такие назначения не помогут полностью избавиться от болезни, но позволят максимально долго обходиться без хирургического вмешательства.
Если мальформация болезни прогрессирующая, тогда понадобится неотложное хирургическое вмешательство. Делается или операция, или шунтирование. Операция позволяет решить две основных причины:
- Исправить дефекты, которые способствуют сдавливанию черепа и головного мозга.
- Приводит в нормальное состояние движение ликвора.
Такая операция довольно распространенная, длительность ее не более двух часов. Пациент восстанавливается полностью через пару недель. Благодаря операции внутричерепное давление нормализуется, а пространство в спинном и головном мозге увеличивается, болезнь отступает.
Профилактические меры
Всегда нужно бережно относиться к своему здоровью, а если это период, когда женщина вынашивает под сердцем тогда ответственность удваивается. Существуют некоторые профилактически меры, позволяющие предотвратить заболевание:
- включить в свой рацион больше фруктов и овощей
- пить свежие соки, кушать молочные изделия и мясо, богатые белком
- пить витамины для беременных
- отказаться от вредных привычек, если таковые имеются
- пить только те препараты, которые допустимы во время беременности и только по назначению врача
- проводить все необходимые обследования
Если следить за своим питанием и вести полноценный, здоровый образ жизни, вовремя сдавать анализы и прислушиваться к врачу, то вероятность родить здорового ребенка увеличивается во много раз.
Итак, синдром Арнольда-Киари у плода возникает по разным причинам, как врожденным, так и приобретенным. Заболевание 1-го и 2-го типа полностью излечимо, если провести необходимую операцию. Чтобы предотвратить появление патологии, будущей маме необходимо максимально позаботиться о своем , тем самым это будет положительно сказываться на развитии головного мозга ее плода.
Фев 25, 2017 Виолетта Лекарь
Мальформация Арнольда–Киари (Arnold-Chiari malformation) — мальформация цервико-медуллярного перехода, характеризуемая смещением миндалин мозжечка и в ряде случаев ствола и IVжелудочка ниже уровня большого затылочного отверстия.
Первым мальформации описал J.Cleland в 1883 г. У 9 умерших младенцев на вскрытии он выявил удлинение ствола и опущение миндалин мозжечка в позвоночный канал. Однако работа J.Cleland не была замечена. Затем в 1891 г. Hans von Chiari описал врожденную аномалию, заключавшуюся в грыжеподобном выпячивании миндалин мозжечка ниже уровня большого затылочного отверстия.
Типы мальформации
Мальформация Киари I типа — смещение миндалин мозжечка вниз через большое затылочное отверстие к верхним отделам спинного мозга. Этот тип мальформации сопровождается гидромиелией и обычно проявляется в подростковом или взрослом возрастах. У подростков главные симптомы — нарушение сгибания и снижение силы в руках, утрата болевой и температурной чувствительности в верхней половине туловища и руках. Взрослые обычно жалуются на боль в шейно-затылочной области, возрастающую при кашле, а также боль в руках.
Мальформация Киари II типа характеризуется смещением червя мозжечка, миндалин, четвертого желудочка и продолговатого мозга (части ствола мозга) в большое затылочное отверстие. Данный тип, называемый также мальформацией Арнольд-Киари, гораздо чаще сопровождается гидромиелией, чем тип I и практически всегда связан с миеломенингоцеле. Миеломенингоцеле — это врожденное нарушение закрытия спинного мозга и позвоночника во время формирования плода. Симптомы этой мальформации очевидны и проявляются обычно сразу после рождения вместе с короткими эпизодами прекращения дыхания, сниженным глоточным рефлексом, непроизвольными и быстрыми движениями глазных яблок вниз, снижением силы в руках.
Мальформация Киари III типа заключается в смещении мозжечка и части ствола мозга с мозговыми оболочками в менингоцеле, расположенное в шейно-затылочной области.
Ранее выделяли мальформацию Киари IV типа, которая сопровождается недоразвитием мозжечка. Однако в настоящее время большинство авторов предпочитает относить этот вид порока развития к Аномалии Денди – Уокера.
Мальформации Арнольда–Киари II и III типов могут сопутствовать признаки дисплазии нервной системы: полимикрогирия, гетеротопия коры, гипоплазия подкорковых узлов, дисгенезия мозолистого тела, патология прозрачной перегородки, утолщение интерталамического соединения, beaking tectum (клювовидный tectum), часто отмечают наличие перегиба сильвиевого водопровода (55%), кисты отверстия Мажанди, гипоплазия серпа и намета мозжечка, hemivertebrae, низкое расположение каудального отдела спинного мозга на уровне LIV–V позвонков и ниже.
Этиология мальформации Киари
Этиология заболевания в настоящее время не ясна. Имеются данные, свидетельствующие о роли генетического фактора в этиологии этого синдрома. Эктопия миндалин мозжечка в затылочное отверстие была обнаружена у трех монозиготных близнецов. После первого описания мальформации Cleland в 1883 г. появилось несколько теорий. Теория, подтверждаемая исследованиями Misao Nishikawa и соавторов, заключается в том, что из-за парааксиальной дисплазии мезодермального листка или первичного повреждения структур соответствующего сомита формируется ненормально маленькая задняя черепная ямка, структуры заднего мозга, заполнив объем задней черепной ямки и продолжая расти, опускаются в затылочный канал. Сочетание Аномалии Киари II типа с менингомиелоцеле связано с тем, что степень парааксиальной дисплазии мезодермального листка при АК – II типа более выражена, чем при АК – I типа и отмечается не только на уровне формирования затылочной кости, но и по оси тела на уровне формирования ряда позвонков, что проявляется в spina bifida, а также в аномалиях ряда других костных структур и костной системы в целом.
Клиническая картина мальформации Киари
Клинические проявления АК – I типа проявляются чаще всего в юношеском либо в зрелом возрасте. Эти проявления укладываются в такие неврологические синдромы, как церебеллобульбарный, ликворогипертензионный, сирингомиелический, синдромы повреждения черепных нервов. Ликворогипертензионный синдром проявляется головной болью, обычно субокципитальной, и болью в шее, усиливающейся при кашле, чихании и напряжении, застойными дисками зрительных нервов. Стволовые нарушения и расстройства функций черепных нервов проявляются в виде неустойчивых осциллопсий, тригеминальной дизестезии, снижения слуха, шума в ушах, головокружения, дисфагии, остановки дыхания во время сна, периодических обмороков (часто связанных с кашлем), нарушения контроля над ЧСС, АД при переходе из горизонтального положения в вертикальное, могут наблюдаться атрофия половины языка, паралич голосовых связок, стридор, спастический или комбинированный (больше в верхних конечностях) тетрапарез. Мозжечковые расстройства - нистагм, дизартрия, атаксия. Симптомы, связанные с сирингомиелическими кистами - онемение, расстройство чувствительности, обычно по диссоциированному типу, а также нейроартропатия, нарушение функций тазовых органов, отсутствие брюшных рефлексов, мышечная гипотрофия. При этом ряд авторов отмечают несоответствие между локализацией, протяженностью кисты, кистозным индексом (отношение переднезаднего размера кисты к таковому размеру поперечника спинного мозга на уровне кисты), с одной стороны, и зоной гипестезии, распространенностью сегментарных расстройств поверхностной чувствительности, выраженностью мышечной гипотрофии и степенью пареза - с другой.
АК II типа манифестирует у новорожденных и в раннем детском возрасте такими симптомами, как апноэ, стридор, билатеральный парез голосовых связок, нейрогенная дисфагия с назальной регургитацией, цианоз во время кормления, нистагм, гипотония, слабость, спастика в верхних конечностях, что может прогрессировать вплоть до тетраплегии.
Мальформация Киари III типа встречается редко, клинические проявления ее такие же, как при АК II.
Диагностика мальформации Киари
Стандартное рентгенологическое исследование может выявить лишь косвенные признаки мальформации АК, компьютерная томография также не дает четкой визуализации мягкотканных структур. Широкое внедрение МРТ в клиническую практику позволило решить большинство проблем, связанных с диагностикой аномалии Киари. Этому способствовали хорошая визуализация структур задней черепной ямки, краниовертебрального перехода, спинного мозга, отсутствие артефактов от костных структур.
Лечение мальформации Киари
Лечение мальформации Киари и сопутствующей сирингомиелии возможно только хирургическим путем. Операция заключается в декомпрессии задней черепной ямки или установке ликворного шунта при сопутствующей гидроцефалии.
Местная декомпрессия выполняется под общей анестезией и состоит в удалении части затылочной кости, а также задних половин I и/или II шейных позвонков до того места, куда спускаются миндалины мозжечка. Опущенные миндалики мозжечка при этом также резецируются, чем достигается устранение компрессии ствола мозга. Эта эффективная операция расширяет большое затылочное отверстие и устраняет сдавление ствола мозга, спинного мозга и миндалин мозжечка. Во время операции открывается также твердая мозговая оболочка — толстая мембрана, окружающая головной и спинной мозг. В раскрытую твердую мозговую оболочку вшивается заплатка из другой ткани (искусственной или взятой у самого больного) для более свободного прохождения ликвора.
Реже выполняются операции по дренированию спинномозговой жидкости из расширенного спинного мозга в грудную или брюшную полость с помощью специального полого шунта с клапаном или в подоболочечное пространство. Иногда эти операции выполняется поэтапно.
Эффективность хирургической декомпрессии составляет от 50 до 85% по данным разных авторов. Необходимо помнить, что хирургическое лечение лучше проводить до развития грубых неврологических нарушений, так как после проведения хирургического лечения восстановление функций происходит не полностью или не происходит вовсе, и основной задачей хирургии является стабилизация неврологического статуса пациента и недопущение дальнейшего прогрессирования заболевания.
Израильский Центр Нейрохирургии и Неврологии Нейромед специализируется на нейрохирургических операциях при аномалии Арнольда-Киари с применением инновационных методик, позволяющих повысить эффективность и безопасность лечения.
Характеризуется несоответствием размеров задней черепной ямки и расположенных в этой области структур мозга, вследствие чего миндалины мозжечка и ствол головного мозга опускаются в большое затылочное отверстие. Это вызывает их ущемление на данном участке. Довольно часто данная патология сочетается с и нарушениями развития спинного мозга.
Большая часть пациентов с синдромом Арнольда-Киари не ощущает никаких симптомов, а обнаруживается заболевание чаще всего случайно, при диагностировании других болезней. Встречается аномалия Киари нечасто: ею страдают примерно 3-8 человек на сотню тысяч случаев.
Каковы причины развития синдрома Арнольда-Киари?
На сегодняшний день этиология этого заболевания до конца не изучена, и единого мнения у специалистов о причинах болезни нет. Существует предположение, что аномалия Арнольда-Киари обусловлена структурными нарушениями в головном и спинном мозге. Развиваются они в период внутриутробного развития и связаны с генетическими мутациями или несбалансированным питанием женщины во время беременности, когда в рационе не хватает питательных веществ.
По другой версии аномалия Киари возникает вследствие увеличенного размера головного мозга, и он как бы вытесняет содержимое задней черепной ямки сквозь затылочное отверстие.
Переход незначительного дефекта в выраженную клиническую форму может спровоцировать гидроцефалия. Она обусловлена увеличением объема мозга за счет увеличения желудочков. Ввиду того, что при синдроме Киари, кроме дисплазии костной ткани краниовертебрального перехода, имеется недоразвитие связочного аппарата на этом участке, то даже малейшая черепно-мозговая травма может усугубить внедрение в затылочное отверстие миндалин мозжечка.
Классификация аномалии Арнольда-Киари
Данная патология подразделяется на четыре типа:
- аномалия Арнольда-Киари I степени - проявляется опущением миндалин мозжечка ниже уровня большого затылочного отверстия. Как правило, проявляется в подростковом и более позднем возрасте. Нередко сопровождается гидромиелией, при которой цереброспинальная жидкость скапливается в спинном мозге, в центральном его канале;
- аномалия Арнольда-Киари II степени - проявляется практически сразу после рождения малыша, в первые дни его жизни. При данной форме заболевания через большое затылочное отверстие, кроме миндалин, выходят продолговатый мозг, червь мозжечка и IV желудочек. Этот тип заболевания часто сочетается с гидромиелией, и практически всегда связан с наличием спинномозговой врожденной грыжей;
- аномалия Арнольда-Киари III степени - проявляется тем, что мозжечок и продолговатый мозг, опускаясь ниже большого затылочного отверстия, располагаются в менингоцеле шейно-затылочного отдела;
- аномалия Арнольда-Киари IV степени - проявляется недоразвитием (гипоплазией) мозжечка, при котором отсутствует его смещение в каудальном направлении. Некоторые специалисты относят данный дефект к синдрому Денди-Уокера, когда кроме гипоплазии мозжечка присутствуют врожденные кисты задней черепной ямки и гиперцифалия.
Аномалии Киари I и II степени довольно часто сочетаются с различными патологиями нервной системы, такими как:
- полимикрогирия;
- гетеротопия коры головного мозга;
- дефекты мозолистого тела;
- кисты отверстия Можанди;
- гипоплазия подкорковых структур;
- перегиб сильвиевого водопровода и др.
Аномалия Арнольда-Киари: симптомы
Симптомы аномалии Арнольда-Киари варьируются в зависимости от типа заболевания. В клинической практике наиболее распространена аномалия Киари I типа, при которой поражаются черепно-мозговые нервы, а также наблюдаются сирингомиелический, церебеллобульбарный и ликворногипертензионный синдромы. Этот вид заболевания выявляется, как правило, уже во взрослом возрасте или в период полового созревания.
Ликворногипертензионный синдром, сопровождающий аномалию Киари I, отличается головной болью в затылочной и шейной областях. Болевые ощущения усиливаются во время напряжения мышц шеи, например, при кашле или чихании. Нередко бывает рвота, которая никак не связана с приемом пищи и ее характером. При осмотре у пациента обнаруживается повышенный тонус шейных мышц. Мозжечковые нарушения проявляются дизартрией (невнятной речью), непроизвольными движениями и расстройством моторики.
Поражение ствола мозга и находящихся в нем корешков и ядер черепно-мозговых нервов вызывают ухудшение зрения, несвязную речь, расстройство глотания, снижение слуха, системное головокружение с ощущением вращающихся вокруг предметов, шумы в ушах, синдром сонных апноэ, повторяющиеся кратковременные потери сознания, ортостатический коллапс. Пациенты отмечают усиление шума в ушах и головокружение при поворотах головы, которые могут спровоцировать даже обморок. Возможны парез гортани и затруднение дыхания.
Аномалия Арнольда-Киари II и III степени сходны в своих клинических проявлениях. Они становятся заметны уже в первые дни жизни малыша. Аномалия Киари II характеризуется шумным дыханием, периодами непродолжительной остановки дыхания, нейропатическим парезом гортани, проблемами с глотанием, при которых жидкая пища забрасывается в нос. У новорожденных наблюдается нистагм, усиление мышечного тонуса, цианоз кожных покровов во время кормления. Могут также присутствовать двигательные расстройства различной степени.
Аномалия Киари III - более тяжелая форма болезни и часто развитие плода не совместимо с жизнью.
Как диагностируется синдром Арнольда-Киари?
При диагностировании данной патологии ни медицинский осмотр, ни набор (РЭГ, ЭЭГ, Эхо-ЭГ) не дают данных, с помощью которых можно поставить точный диагноз. Они позволяют выявить только признаки гидроцефалии.
Рентгенографическое исследование определяет только костные дефекты, сопровождающие аномалию Киари. До внедрения томографии в неврологическую практику диагностика данного заболевания была затруднительна. Теперь возможна постановка точного диагноза.
Синдром Арнольда-Киари: лечение
Как уже отмечалось выше, заболевание в большинстве случаев протекает без каких-либо признаков, поэтому очень часто синдром Арнольда-Киари лечение не предполагает. При наличии болей в шейной и затылочной областях проводят консервативную терапию. Она включает анальгетики, миорелаксирующие и противовоспалительные препараты.
Неврологические нарушения (расстройства мышечного тонуса и чувствительности, парезы и пр.), а также при неэффективности лечения болевых синдромов консервативным методом применяется хирургическое вмешательство, цель которого - остановить процесс структурных изменений в позвоночнике и мозге, а также стабилизировать симптомы. При удачном исходе операции давление на мозжечок снижается, и отток ликвора восстанавливается.
Существуют заболевания, связанные с аномальным расположением органов из-за чего, возникают дисфункции на этих участках. Одним из таких недугов является синдром Арнольда Киари (мальформация Киари). Для него свойственно аномальное расположение мозжечка и ствола головного мозга в черепной коробке из-за чего они немного выходят в затылочное отверстие. Такое явление сопровождается болевыми приступами в области затылка, бессвязной речью, нарушением координации, ослаблением мышц гортани и другими симптомами. Делится заболевание на 4 разновидности, каждая из которых имеет свои признаки и лечение.
Аномалия Киари диагностируется достаточно просто и для этого хватит сделать МРТ. Вылечить патологию можно только с помощью хирургического вмешательства.
Между спинным и головным мозгом есть определенное разграничение, которое представляет собой затылочное отверстие. В этом месте они соединяются и над ним локализована задняя черепная ямка, содержащая мозжечок, мост и продолговатый мозг. При Киари болезни в затылочное отверстие выпадают мозговые ткани из-за чего сдавливается участки продолговатого и спинного мозга, локализованные в этом участке. Часто из-за такого процесса ухудшается отток ликвора из головы (спинномозговой жидкости), что влечет за собой развитие гидроцефалии (водянки головного мозга).
Относится синдром Киари к врожденным патологиям соединения черепа и верхнего отдела позвоночника.
Ставится такой диагноз достаточно редко и по статистике он наблюдается у 10 новорожденных на 120 тыс. человек.
Выявить наличие болезни можно через один-два дня с момента появления на свет, но некоторые типы заболевания обнаруживаются лишь во взрослом возрасте. Чаще всего болезнь Арнольда Киари развивается вместе с сирингомиелией, для которой свойственны пустые участки в веществе спинного и продолговатого мозга.
Причины развития
Аномалия Арнольда Киари до сих пор особо не изучена и точных факторов, которые влияют на развитие болезни никто не может назвать. Существует множество предположений, например, некоторые специалисты считают, что патология вызывается из-за чересчур маленького размера задней черепной ямки. Вследствие этого тканям не хватает места, и они опускаются в затылочное отверстие. Альтернативная теория гласит в том, что головной мозг с рождения больше обычного размера. Именно поэтому он выталкивает часть своих тканей в затылочное отверстие.
Вследствие застоя ликвора и последующего формирования гидроцефалии патология имеет более выраженные симптомы. Ведь из-за этой болезни увеличиваются желудочки, а вследствие и размер мозга. Такой процесс усиливает выдавливание миндалин мозжечка в затылочное отверстие. Усугубить течение болезни могут и травмы головы. Для болезни Киари свойственно не только неправильное развитие тканей перехода между спинным и головным мозгом, но и аномалии связочного аппарата. Любое повреждение в области головы вдавливает еще больше мозжечок в затылочную область из-за чего болезнь проявляется сильнее.
Разновидности патологического процесса
Мальформация Киари имеет такие типы развития:
- Первый вид. Для аномалии Арнольда Киари 1 типа свойственно попадание миндалин мозжечка в затылочное отверстие. В основном проявляется болезнь в подростковом периоде, а иногда уже во взрослом возрасте. Крайне часто при мальформации Арнольда Киари 1 типа возникает гидромиелия;
- Второй вид. Этот диагноз ставится уже в первые дни с момента появления ребенка на свет. Аномалия Арнольда Киари 2 типа протекает значительно тяжелее 1 вида. В ее случаи в затылочную впадину попадает еще и кусочек мозжечка (помимо миндалины), а также четвертый желудочек и продолговатый мозг. Гидроцефалия при этом типе болезни возникает значительно чаще. Причина у этого явления в большинстве случаев кроется во врожденной ;
- Третий вид. Его отличия от второго типа состоят в том, что опустившиеся в затылочное отверстие ткани попадают в менингоцеле (грыжу), которая расположена в шейно-затылочной части;
- Четвертый вид. Его суть в недоразвитом мозжечке, который в отличие от других типов болезни не смещается в затылочное отверстие. Некоторые специалисты считают, что болезнь Киари 4 типа является частью синдрома Денди-Уокера. Для него свойственно развитие кист, локализованных в области задней черепной ямки и водянки головного мозга.
Вторая и третья разновидность аномалии часто идет в комплексе с неправильным развитием других тканей нервной системы, а именно:
- Атипичным расположением тканей коры головного мозга;
- Патологиями мозолистого тела;
- Полимикрогирией (множество маленьких извилин);
- Недоразвитостью тканей подкорковых структур, а также серпа мозжечка.
Симптоматика
Аномалия Арнольда Киари 1 степени диагностируется чаще всего. Она сочетает в себе такие синдромы:
- Сирингомиелический;
- Ликворногипертензионный;
- Церебеллобульбарный.
На этом фоне поражаются черепно-мозговые нервные волокна, а проживают без симптомов с этой патологией вплоть до подросткового возраста. У некоторых людей первые признаки становятся видны уже после 20 лет.
Ликворногипертензионный синдром, который характерен для аномалии Арнольда Киари, проявляется такими признаками:
- Болевые ощущения в шейной и затылочной области, которая проявляется вовремя кашля и чихания, а также из-за напряжения мышц в этом месте;
- Беспричинная рвота;
- Напряжение мышц шеи;
- Бессвязная речь;
- Нарушенная координация движений;
- Неконтролируемое колебание глаз (нистагм).
Со временем из-за повреждения ствола головного мозга и окружающих его нервов у человека наблюдаются такие симптомы:
- Ухудшение зрения;
- Раздвоенная картинка перед глазами (диплопия);
- Проблемы с глотанием;
- Ухудшение слуха;
- Частое головокружение;
- Шум в ушах;
- Нарушение сна сопровождающиеся остановкой дыхания через нос и рот на 10 и более секунд;
- Потеря сознания;
- Недостаточное кровоснабжение головного мозга, проявляющиеся из-за низкого артериального давления при смене положения тела.
При аномалии Арнольда Киари симптомы усиливаются из-за резких поворотов головы и болезнь проявляется следующим образом:
- Усиливается головокружение;
- Становится громче шум в ушах;
- Чаще теряется сознание;
- Половина языка подвергается атрофическим изменением (уменьшению в размере);
- Ослабевают мышцы гортани из-за чего становится трудно дышать, а хрипнет голос;
- Ослабление мышц конечностей, преимущественно верхних.
Зачастую патологии свойственен сирингомиелический синдром и в таком случае она проявляется следующим образом:
- Ухудшение чувствительности;
- Белково-энергетическая недостаточность (гипотрофия) мышечной ткани;
- Онемение;
- Ослабевание или полное отсутствие брюшных рефлексов;
- Тазовые нарушения;
- Нейрогенная артропатии (деформация сустава).
Церебеллобульбарный синдром, возникающий при болезни Киари, проявляется такими симптомами:
- Нарушение функций тройничного нерва;
- Вестибулокохлеарный нервоз;
- Проблемы с координацией;
- Нистагм;
- Головокружение.
Вторая и третья степень болезни проявляется похожим образом, но первые симптомы видны уже через 2-3 дня с момента рождения. Для 2 типа аномалии Арнольда Киари есть свои отличительные признаки:
- Громкое дыхание, которое иногда останавливается на 10-15 секунд;
- Двухсторонние ослабление мышц гортани, вследствие чего развиваются проблемы с глотанием, и жидкая пища часто забрасывается в нос;
- Нистагм;
- Затвердевание мышц верхних конечностей из-за повышенного тонуса;
- Посинение кожного покрова (цианоз);
- Затруднение движений вплоть до паралича большей части тела (тетраплегии).
Третий вид аномалии протекает значительно тяжелее и редко совмести с жизнью из-за серьезных нарушений.
Диагностика
В былые времена было крайне тяжело диагностировать патологию, так как опрос, осмотр и стандартные методы обследования не давали особых результатов. Они показывали наличие повышенного давления и водянки головного мозга. Рентген немного упрощал задачу, так как на нем были видны костные деформации в черепе, но это не позволяло полностью быть уверенным в диагнозе. Ситуация изменилась после появления топографических исследований. Ведь такой метод диагностики позволял полностью рассмотреть образования на задней черепной ямки. Именно поэтому МРТ (магнитно-резонансная томография) считается незаменимым методом обследования для дифференцирования синдрома Киари среди других патологических процессов.
С помощью МРТ потребуется обследовать область шеи и груди. Ведь в этих местах позвоночника часто находятся менингоцеле, а также сирингомиелические кисты. Во время проведения обследования врач должен не только убедиться в наличии синдрома Киари, но и исключить другие патологические процессы, которые часто с ним сочетаются.
Курс терапии
Для аномалии Арнольда Киари лечение не требуется только в том случае, если патология протекает бессимптомно. В такой ситуации следует избегать ударов и повреждений в области головы и шеи, чтобы не усугубить течение болезни.
Если заболевание протекает с минимальной симптоматикой, а именно со слабовыраженной болью, то необходим курс медикаментов с обезболивающим и противовоспалительным эффектом. Не помешает назначение миорелаксантов для расслабления мышц.
В тяжелых случаях, когда болезнь протекает с ярко выраженной симптоматикой (ослабление мышц, дисфункции черепно-мозговых нервов и т. д.) потребуется хирургическое вмешательство. Во время выполнения операции врач расширит затылочное отверстие, с помощью удаления кусочка затылочной кости. Если нужно убрать давление со ствола и спинного мозга, то потребуется удалить часть миндалин мозжечка и переднюю половину 2 верхних позвонков. Чтобы нормализировать циркуляцию ликвора хирургу потребуется сделать заплатку в твердую мозговую оболочку.
В некоторых случаях лечение хирургическим путем проводится с помощью шунтирования. Такая операция представляет собой ответвление ликвора с помощью дренажа вследствие чего он перестает застаиваться.
Прогноз

Многие люди, страдающие от синдрома Арнольда Киари, задаются вопросом сколько им осталось жить. Ответить на этот вопрос можно опираясь на вид патологии и тяжесть течения. Важным фактором также является своевременность хирургического вмешательства.
Люди, страдающие от болезни Киари 1 типа, часто имеют среднестатистическую продолжительность жизни, так как патология может протекать бессимптомно. Если 1 или 2 вид заболевания имеют неврологические признаки, то важно по скорее выполнить операцию. Ведь осложнения, связанные с головным и спинным мозгом, фактически не поддаются лечению. Третий тип болезни чаще всего заканчивается смертью больного при рождении.
Болезнь Арнольда Киари является врожденной аномалией, которую необходимо лечить при проявлении первых симптомов. В ином случае можно навсегда остаться инвалидом или лишиться жизни.
Аномалия Арнольда-Киари – это врожденная патология развития ромбовидного мозга, проявляющаяся несоответствием размеров задней черепной ямки и мозговых структур находящихся в этой области, что приводит к опущению ствола головного мозга и миндалин мозжечка в большое затылочное отверстие и ущемлению их на этом уровне.
В большинстве случаев дефект сочетается с гидроцефалией и аномалиями развития спинного мозга. Причинами могут быть врождённая дисплазия (нарушение) широкого затылочного отверстия, размеры которого становятся значительно больше нормы.
Впервые он был описан Н. Chiari в 1896 г. Это состояние характеризуется каудальным смещением продолговатого мозга, моста и червя мозжечка, когда все эти структуры оказываются в шейной части позвоночника.
Частота этого заболевания составляет от 3.3 до 8.2 наблюдений на 100000 населения.
Истинная частота различных типов синдрома Арнольда - Киари, да и частота этого порока в целом, не установлены. Одной из причин отсутствия таких данных являются разные подходы к классификации этого порока. Согласно Международной классификации болезней, синдром Арнольда - Киари имеет отдельный шифр (Q07.0), однако определяется в ней как «... патологическое состояние, при котором происходит повышение внутричерепного давления в результате интракраниальной опухоли, окклюзионных форм гидроцефалии, воспалительного процесса, что в некоторых случаях приводит к вклинению мозжечка и продолговатого мозга в большое затылочное отверстие». В ультразвуковой пренатальной литературе до сих пор не удалось найти описаний случаев дородовой диагностики синдрома Арнольда- Киари, полностью соответствующих этим характеристикам.
Морфологические особенности различных типов порока Арольда - Киари определяют возможности пренатального выявления и прогноз для жизни.
Причины возникновения синдрома Арнольда- Киари до конца не установлены. Хромосомные аномалии при этой патологии, как правило, выявить не удается.
Патогенез (что происходит?) во время Аномалии Арнольда-Киари:
До настоящего времени патогенез патологии окончательно не установлен. По всей вероятности, этих патогенетических факторов три:
первый - наследственно обусловленные врожденные остеоневропатии,
второй - травматические повреждения клиновидно-решетчатой и клиновидно-затылочной части ската вследствие родовой травмы,
третий - гидродинамический удар ликвора в стенки центрального канала спинного мозга.
Анатомические особенности аномалии Киари
Мозжечок расположен в задней черепной ямке. (ЗЧЯ)
Миндалины - это нижняя часть мозжечка. В норме они расположены выше большого затылочного отверстия. При аномалии Киари миндалины мозжечка находятся ниже большого затылочного отверстия, в позвоночном канале.
Большое затылочное отверстие - это своеобразная граница между черепом и позвоночником, между головным и спинным мозгом. Выше большого затылочного отверстия находится задняя черепная ямка, ниже позвоночный канал.
На уровне большое затылочного отверстия нижний отдел ствола мозга (продолговатый мозг) переходит в спинной мозг. В норме спино-мозговая жидкость (ликвор) свободно циркулирует в субарахноидальных пространствах головного и спинного мозга. На уровне большого затылочного отверстия субарахноидальные пространства головного и спинного мозга соединяются, что обеспечивает свободный отток ликвора от головного мозга.
При аномалии Киари низко расположенные миндалины мозжечка затрудняют свободную циркуляцию спиномозговой жидкости между головным и спинным мозгом. Миндалины блокируют большое затылочное отверстие, как пробка затыкает бутылочное горлышко. В результате нарушается отток ликвора и развивается гидроцефалия.
Симптомы Аномалии Арнольда-Киари:
Киари (Chiari) выделил четыре типа аномалии с подробным их представлением. Данной классификацией врачи пользуются по настоящее время.
1. Аномалия Арнольда-Киари I типа представляет собой опущение структур ЗЧЯ в позвоночный канал ниже плоскости большого затылочного отверстия.
2. При аномалии Арнольда-Киари II типа - происходит каудальная дислокация нижних отделов червя, продолговатого мозга и IV желудочка, нередко развивается гидроцефалия.
3. Аномалия Арнольда-Киари III типа встречается редко, характеризуется грубым каудальным смещением всех структур задней черепной ямки.
4. Аномалия Арнольда-Киари IV типа - гипоплазия мозжечка без смещения его вниз.
Аномалии III и IV типов обычно несовместимы с жизнью.
Примерно у 80% пациентов аномалия Арнольда-Киари сочетается с патологией спинного мозга – сирингомиелией, которая характеризуется образованием в спинном мозге кист, вызывающих прогрессирующую миелопатию. Эти кисты образуются при опущении структур задней черепной ямки и сдавление шейного отдела спинного мозга.
Типичная клиническая картина аномалии Арнольда-Киари характеризуется следующими симптомами :
Боль в шейно-затылочной области усиливающаяся при кашле, чихании,
Снижение болевой и температурной чувствительности в верхних конечностях,
Снижение мышечной силы в верхних конечностях,
Спастичность верхних и нижних конечностей,
Обмороки, головокружения,
Снижение остроты зрения,
В более запущенных случаях присоединяются: эпизоды апноэ (короткая остановка дыхания), ослабление глоточного рефлекса, непроизвольные быстрые движения глаз.
Возможные последствия, осложнения:
1. На фоне нарастающих признаков внутричерепной гипертензии (иногда без нее) отмечаются прогрессирующие нарушения функции мозжечка и сдавление шейного отдела спинного мозга, параличи черепных нервов.
2. Иногда аномалия Арнольда-Киари сочетается и с костными дефектами - окципитализацией атланта и базилярной импрессией (воронкообразное вдавление ската и краниоспинального сочленения).
3. Аномалии позвоночника, деформации стоп.
Диагностика Аномалии Арнольда-Киари :
Иногда аномалия Киари никак не проявляет себя и выявляется случайно при диагностических процедурах.
В настоящий момент методом выбора при диагностике данной патологии является МРТ головного мозга шейного и грудного отделов спинного мозга (для исключения сирингомиелии).
Лечение Аномалии Арнольда-Киари :
Если единственным симптомом заболевания является незначительной интенсивности болевой синдром, для лечения применяется консервативная терапия, которая включает в себя различные схемы с примененим нестероидных противовоспалительных препаратов и миорелаксантов.
При отсутствии эффекта от консервативной терапии в течение 2-3 месяцев или наличии у пациента неврологического дефицита (онемение, слабость в конечностях и т.д.) показано проведение операции.
Целью операции является – ламинэктомия, декомпрессивная краниоэктомия задней черепной ямки и пластика твёрдой мозговой оболочки. При подобной операции увеличивается объём задней черепной ямки и расширение затылочного отверстия, в результате чего прекращается сдавление нервных структур и нормализация тока цереброспинальной жидкости. В случаях сопуствующей гидроцефалии выполняется шунтирующая операция.
В Израиле больным предлагается щадящее и качественное лечение, которое после лечения позволяет больным вести полноценную жизнь. Хирургическое лечение синдрома Арнольда-Киари выполняется при помощи эндоскопа, при этом травмирующий эффект хирургического лечения сводится к минимуму. Метод минимально инвазивного хирургического лечения проводимый в израильских клиниках дает возможность пациентам с аномалией Арнольда-Киари вести в последующем полноценный образ жизни даже без лекарственной поддержки.
Симптоматология синдрома Арнольда-Киари
Костноcyставные врожденные аномалии клинически менее очевидны сами по себе и гораздо более вследствие их тяжелых осложнений на центральной и периферической нервной системе. Неврологические проявления переносятся больным тяжелее всего и обусловливают неблагоприятное течение этого синдрома. Вообще, начало болезни медленное и нехарактерное.
Симптоматология, вначале очень стертая и даже отсутствующая долгое время, часто выявляется в результате вмешательства ряда разрешающих факторов, как, например, черепно-мозговая травма или инфекции носоглотки.
Первым проявлением, привлекающим внимание на наличие врожденной аномалии непосредственно после родов, является наличие миеломенингоцеле (спинномозговая грыжа). Позже отмечаются и другие клинические явления, указывающие на наличие костносустваных и неврологических аномалий, а именно:
- расщепление позвоночника,
- латеральное наклонение головы,
- девиация глазных яблок,
- головная боль и прерывистые или проходящие боли в области шеи (в особенности у детей старшего возраста и у взрослых), появляющиеся при движениях головы;
- тошнота, рвота.
У многих больных из-за блокады циркуляции спинномозговой жидкости между 4-м желудочком и цистернами основания черепа, уже в течение первых месяцев жизни, развивается иволютивная внутренняя гидроцефалия, обусловливающая появление очень многих и различных неврологических явлений. Постепенно череп ребенка увеличивается в размере и появляется затруднение в связи с кормлением, а также и дыхательные расстройства, а менингоцеле (когда оно существует) может изъязвиться.
Проявления внутричерепной гипертонии:
- сильные головные боли,
- папиллярный застой или атрофия зрительного нерва (поздняя),
- сопровождаемая прогрессивными нарушениями зрительной функции, вплоть до полной слепоты.
Мозжечковые проявления:
- головокружение;
- атаксия при ходьбе и в ортостатическом положении;
- дизартрия;
- нарушение глотания,
- интенционное дрожание,
- нистагм.
Проявления в сфере периферической нервной системы:
- парестезии, анестезии, парезы или параличи спастического типа,
- усиленные костносухожильные рефлексы,
- наличие рефлекса Бабинского.
Проявления в сфере черепно-мозговых нервов:
- односторонний или, реже, двусторонний паралич лицевого нерва;
- паралич глазодвигательных нервов, выраженный чаще всего внутренним косоглазием или диплопией.
Диагностика синдрома Арнольда-Киари.
Люмбальная пункция и биохимический и бактериологический анализы спинномозговой жидкости в большинстве случаев не доставляют значительных данных. Кроме того, применение люмбальной пункции может ухудшить состояние больного и даже вызвать смертельный исход ввиду внезапного понижения давления и полного проникновения мозжечковых миндалин и продолговатого мозга в позвоночный канал.
Обыкновенное рентгенологическое обследование выявляет следующие аспекты: малая задняя черепная ямка; расширение затылочного отверстия и позвоночного канала; гидроцефалия (большой череп с расхождением швов); отпечатки пальцев на костной пластинке черепа; сплющенность турецкого седла; шейное, дорзальное и люмбальное расщепление позвоночника.
Проведение дополнительных рентгенологических обследований (газовая миелоэнцефалография) противопоказаны у детей моложе 2 лет. Однако у детей старшего возраста и у взрослых они определяют косвенные и прямые признаки гидроцефалии, смещение продолговатого мозга и миндалин мозжечка, а также и сдавливание спинного мозга в шейной области.
Патологоанатомическое исследование. С анатомической и топографической точек зрения синдром Арнольда – Киари существует в виде четырех хорошо индивидуализированных типов, а именно:
- Первый тип, при котором существует растягивание и опущение миндалин мозжечка без смещения продолговатого мозга. Этот тип встречается чаще всего у детей старшего возраста и у взрослых. С клинической точки зрения он может оставаться всю жизнь бессимптомным и его выявление может произойти совершенно случайно.
- Второй тип, при котором смещается нижняя часть мозжечка и продолговатого мозга через большое затылочное отверстие в позвоночный канал. Этот тип встречается чаще у грудных детей и клинически проявляется гидроцефалией и зачастую и наличием миеломенингоцеле.
- Третий тип, с полным проникновением мозжечка в миеломенингоцеле шейных позвонков.
- Четвертый тип, при котором отмечается гипоплазия мозжечка, вызванная его тотальной грыжей; червь мозжечка невозможно различить, а миндалины и клочок мозжечка - едва заметны. Подобная форма встречается исключительно редко.
Помимо аномалии затылочной кости и позвонков, затрагивающих мозжечок и продолговатый мозг, существуют и другие черепно – позвоночные аномалии, а именно спаивание первого позвонка с затылочной костью, продвижение вверх позвоночника в черепную область из-за гипоплазии затылочной кости; слияние двух-трех позвонков (чаще всего 2-го и 3-го шейных позвонков), как это происходит при синдроме Klippel-Feil; шейное, дорсальное или люмбальное расщепление позвоночника.
Течение и прогноз синдрома Арнольда-Киари . Течение заболевания медленное. Появление синдрома у новорожденного несовместимо с его выживаемостью и его течение ведет быстро к летальному исходу.
В случае более медленного течения, синдром Арнольда – Киари осложняется появлением хронического арахноидита и паренхиматозными поражениями аксона, особо значимым это становится, когда наступает расстройство кровообращения в нервной ткани. Обычно, при подобных случаях нейропсихические расстройства отмечаются поздно и проявляются в форме параплегии, тетраплегии и запозданием психического развития.
Лечение синдрома Арнольда-Киари . Единственным аффективным лечением является хирургическое вмешательство. Показания к операции не ставятся на основании рентгенологического определения костных аномалий, а только тогда, когда последние сопровождаются тяжелыми неврологическими проявлениями. Хирургическое вмешательство заключается в затылочной краниотомии в сочетании с высокой ламинэктомией; твердая оболочка рассекается и оставляется открытой. Фиброзные спайки, часто обширные, существующие вокруг затылочного отверстия, пересекаются и отслаиваются.
При наличии выраженной гидроцефалии, производится деривация желудочка при помощи клапана Holter или Pudenz (по классическому методу хирургического вмешательства по поводу гидроцефалии).
Хотя в некоторых случаях, немедленные результаты и являются благоприятными, все же, все хирургические вмешательства при синдроме Арнольда – Киари связаны с большим послеоперационным риском, вследствие нарушений в области продолговатого мозга, которые могут наступить в ближайший послеоперационный период и вызывать часто смертельный исход.

