НАСЛЕДСТВЕННЫЕ ЗАБОЛЕВАНИЯ НЕРВНОЙ СИСТЕМЫ
ЛЕКЦИЯ 16
Дегенеративные заболевания с преимущественным поражением нервно-мышечного аппарата составляют самую значительную группу среди всех наследственных заболеваний.
Исключительно важными, а нередко решающими в диагностике нервно-мышечных заболеваний являются результаты электрофизиологических и биохимических исследований. Столь же велико значение патоморфологических находок. Изучение мышечного биоптата в световом микроскопе помогает дифференцировать миогенную атрофию от неврогенной. Гистохимическое исследование необходимо для выявления метаболических поражений мышц, а электронная микроскопия открыла целый большой класс заболеваний - непрогрессирующие миопатии.
Осторожное растяжение пораженных суставов уменьшает возникновение контрактур и, таким образом, приводит к более медленному прогрессу заболевания. В этом случае выполняются различные конкретные упражнения, а повязка давления Уриаса оказалась эффективной в случае нарушенной глубины конечностей и часто считается домашней обработкой при выписке. Сенсомоторная тренировка пациентов с мышечными заболеваниями проводится в индивидуальной или групповой терапии и фокусируется на координации и тонких моторных навыках.
Очень важным методом лечения людей с заболеваниями мышц является восстановление способности снова водить автомобиль. Для этой цели существует сотрудничество с внешней автошколой в отделении профессиональной терапии, а вождение в Бад-Вильдунгене проводится в подходящем автомобиле с инвалидностью с обученным инструктором по вождению.
Прогрессирующие мышечные дистрофии. Термином мышечные дистрофии называется группа генетически детерминированных расстройств, характеризующихся прогрессирующими дегенеративными изменениями в мышечных волокнах без первичной патологии периферического (нижнего) мотонейрона.
Различные формы отличаются друг от друга типами наследования, сроками начала процесса, характером и быстротой его течения, своеобразием топографии мышечных атрофии, наличием или отсутствием псевдогипертрофий и сухожильных ретракций и другими признаками.
Дополнительным подходом в реабилитации мышечных заболеваний является предоставление адекватного консультирования. Это относится не только к ортезам, но и ко всем другим вспомогательным средствам дома, Целенаправленная помощь инвалидам-инвалидам с мышечными заболеваниями чрезвычайно полезна, инвалидные кресла точно измерены и приспособлены к потребностям людей, страдающих нарушениями мышц. Душевое кресло, подъемник для ванной, инвалидные кресла, пандусы для инвалидных колясок и другие.
Отделение физической терапии использует различные процедуры для расслабления пациента. Кроме того, этот отдел лечения может уменьшить часто незначительные болевые симптомы у пациентов с нарушениями мышц. Речевая терапия - это еще один отдел реабилитации для лечения мышечных заболеваний. Здесь также должен быть проведен еще один диагноз с ларингоскопией или определенными рентгенологическими исследованиями. В квалифицированной реабилитационной клинике для лечения мышечных заболеваний целостный терапевтический подход к этому заболеванию является важной предпосылкой.
Большинство мышечных дистрофий хорошо изучено клинически, их подробное описание сделано еще в конце прошлого века. Но, несмотря на почти вековую историю изучения миодистрофий, вопросыих патогенеза и лечения остаются до сего времени неразрешенными. Большие надежды возлагаются на молекулярную генетику, с помощью которой определено местонахождение генов уже многих нозологических форм.
Сюда входят аспиранты-психологи факультета. Затрагиваемые лица страдают из-за физического дефицита и связанных с ним последствий в частно - Продолжительность жизни часто связана с депрессивными настроениями и психосоматическими жалобами. Здесь очень полезны поддерживающие отдельные беседы или изучение релаксационных методов, таких как аутогенное обучение или прогрессирующая релаксация мышц.
Вопросы, касающиеся закона о сестринском деле, строгого инвалидности, рабочего места, контактов с работодателем для профессиональной реабилитации или внедрения в компании, должны упоминаться здесь как частые темы в социальном консультировании. Кроме того, целевое консультирование при консультациях часто является частым вопросом для пациентов с нарушениями мышц. Наконец, план терапии во время реабилитации в Бад-Вильдунгене также включает координацию квалифицированного ортопедического специалиста. Вицерек является главным врачом ортопедического отделения клиники в Хомберге в Бад-Вильдунгене.
Диагностика мышечных дистрофий нередко представляет большие трудности. Имеется большая вариабельность клинических проявлений, а малое число членов семьи затрудняет определение типа наследования.
Характерным моторным дефектом у больных с мышечными дистрофиями является “утиная” походка: больной ходит переваливаясь к боку на бок. Она связана главным образом со слабостью ягодичных мышц, прежде всего средней и малой, которые фиксируют таз относительно бедренной кости. В результате при заболевании возникает наклон таза в сторону неопорной ноги (феномен Тренделенбурга) и компенсаторный наклон туловища в противоположную сторону (феномен Дюшенна). При ходьбе сторона наклона постоянно меняется. Указанные изменения можно проверить и в пробе Тренделенбурга, попросив больного поднять одну ногу, согнув ее под прямым углом в коленном и тазобедренном суставах: таз на стороне поднятой ноги опускается (а не поднимаетсякак в норме) из-за слабости средней ягодичной мышцы опорной ноги.
В клинике проводятся регулярные тренинги, а также участие в соответствующих конгрессах и симпозиумах. Эта встреча состоялась, в частности. Часто также наблюдается увеличение мышечных ферментов. Болезни обычно развиваются медленно ползучая, но происходят быстрые болезни. Если вы подозреваете, что у вас миопатия или участвуете в известном нейромышечном заболевании, вы можете назначать встречи.
Лечение зависит от конкретной причины, воспалительное мышечное заболевание может, например, Например, различными иммунотерапевтическими средствами. Подробный обзор разнообразия генетических мышечных расстройств можно найти в гена-таблицах нервно-мышечных заболеваний.
Поднимаясь из горизонтального положения, больной с выраженной мышечной слабостью проксимальных мышц с трудом переворачивается на живот, затем, упираясь руками в пол, становится на четвереньки и после этого, упираясь руками в голени, затем в бедра, постепенно выпрямляется. Этот феномен “набирания по себе” носит название приема Говерса. Часто он связан со слабостью больших ягодичных мышц.
Лекарства, которые могут вызвать или увеличить мышечную болезнь
В нашей практике доступны все необходимые диагностические возможности. В этом участвует практика, организована регулярная тренировка в области нервно-мышечных заболеваний. Затронутые лица также могут зарегистрироваться в регистре пациентов. Реестры пациентов являются важным первым шагом на пути к лучшему пониманию болезней и исследований в области развития терапии.
Сердечное поражение мышц
О возможном вовлечении сердечной мышцы в миопатии и других нервно-мышечных заболеваниях обзор в этом.Миодистрофия Дюшенна. Псевдогипертрофическая мышечная дистрофия Дюшенна встречается чаще всех других заболеваний мышечной системы (30 на 100000 живых новорожденных). Характеризуется ранним началом и злокачественным течением. Классическая картина проявляется изменением походки у ребенка в возрасте 2-5 лет, к 8-10 годам дети ходят уже с трудом, к 14-15 годам они, как правило, полностью обездвижены. У детей более раннего возраста начальные симптомы проявляются отставанием двигательного развития: они позднее начинают ходить, не умеют бегать и прыгать. Больные умирают на 2-3-м десятилетии жизни.
Мы имеем в виду все нервно-мышечные заболевания. Согласно классификации Уолтона, существует 800 форм. Этот симтом может быть ограничен несколькими группами мышц, или, в зависимости от заболевания, вся мускулатура может быть обнаружена. Мышечное истощение, уменьшение мышечной массы и мышечная слабость являются основными симптомами нервно-мышечных заболеваний. Лекарства называют снижение мышечной мышечной атрофии мышц. Тем не менее, они еще не обеспечивают прямой диагноз. Существует ряд причин, которые могут привести к частичному уменьшению массы мышцы, что частично связано с целым телом.
Одними из первых признаков заболевания являются уплотнения икроножных мышц и постепенное увеличениеихобъема за счет псевдогипертрофий. Атрофии мышц бедра, тазового пояса нередко маскируются хорошо развитой подкожной жировой клетчаткой. Постепенно процесс принимает восходящее направление и распространяется за плечевой пояс, мышцы спины, а затем и на проксимальные отделы рук.
Реальная причина такой мускульной атрофии или мышечной слабости может лежать в отдаленных, совершенно разных областях тела. Причины могут быть в нервных клетках спинного мозга, ответственных за движение, в питающих нервах, при переносе нерва в мышцу или в самих мышцах.
Лечение нервно-мышечных заболеваний
Существуют многообещающие препараты для лечения миозитов, миастении и эндокринной миопатии. Первые подходы к замедлению возможны при боковом склерозе амиотрофики. В случае наследственных мышечных дистрофий и спинальных мышечных атрофий каузативное лечение еще не установлено. Особые ожидания на будущее направлены на генную терапию. Во всех нервно-мышечных заболеваниях симптоматика представляет собой последовательную физиотерапевтическую поддержку в сочетании с ортопедическими мерами, в ряде случаев с поддержкой дыхания и в отдельных случаях с возможностью трансплантации сердца.
В терминальной стадии слабость мышц может распространяться на мышцы лица, глотки, дыхательные мышцы.
В развитой стадии болезни имеются такие характерные симптомы, как “утиная походка”; подчеркнутый поясничный лордоз, крыловидные лопатки, симптом “свободных надплечий”. Типичны ранние мышечные контрактуры и сухожильные ретракции, особенно ахилловых сухожилий. Рано выпадают коленные рефлексы, а затем рефлексы с верхних конечностей.
Лечение также включает компенсацию физических ограничений с помощью приспособленных средств. Немецкое общество мускульной болезни создало независимый ресурсный центр. Существует также возможность протестировать средства в домашней обстановке в двух типовых квартирах или провести всего несколько дней в безбарьерных квартирах.
Мышечные расстройства Независимые расстройства Мышечные относительно редко. С другой стороны, часто случается, что на мышцы влияют другие основные заболевания, особенно болезни нервной системы; Инфекционные болезни, такие как тиф и туберкулез; Паразитарные заболевания, Троицы и свиные черви, а также гормональные расстройства.
Псевдогипертрофии могут развиваться не только в икроножных, но также в ягодичных, дельтовидных мышцах, мышцах живота, языка. Очень часто страдает сердечная мышца по типу кардиомиопатии. Выявляются нарушения ритма сердечной деятельности, расширение границ сердца, глухость тонов, изменения ЭКГ. Острая сердечная недостаточность - наиболее частая причина летальных исходов при миодистрофии Дюшенна. На вскрытии находят фиброз и жировую инфильтрацию сердечной мышцы.
Простая мышечная потеря часто встречается в результате сохранения или отдыха, Сокращение мышц может быть вызвано спайками или травмами из-за плохой циркуляции - В результате удушения ассоциаций. Причиной боли в мышцах может быть ревматизм мышц, перенапряжение, холод, нарушение обмена веществ или перенапряжение определенных групп мышц из-за деформации скелета.
Миома - доброкачественная опухоль мышечной ткани. Полимиозит: Мышечное заболевание с аналогичными симптомами, как при дерматомиозите, но без кожной сыпи. Что касается причины, то предполагается, что нарушение относится к аутоагрессиям, как к страданиям, которые приходят в результате нападения тела на собственные структуры.
Нередко наблюдается нарушение моторики желудочно-кишечного тракта.
Частым симптомом является снижение интеллекта. Представляет интерес тот факт, что в одних семьях олигофрения бывает резко выражена, в других сравнительно умеренно. Изменение высших психических функций обычно не прогрессирует и не коррелирует с тяжестью мышечного дефекта. Оно не может быть объяснено только педагогической запущенностью больных детей, которые рано выключаются из детских коллективов, не посещают детский сад и школу из-за двигательных дефектов. При КТ и МРТ нередко обнаруживают церебральную атрофию, возможно, связанную с нарушением пренатального развития головного мозга.
Существует более 200 форм мышечных заболеваний, некоторые из которых были названы в честь их первооткрывателей, а другие после расстройства. Наиболее распространены три подгруппы. Прогрессирующая мышечная дистрофия Спинная мышечная атрофия Нейронная мышечная атрофия. В клинической практике эти заболевания редки и абсолютно необходимы в руках специалиста-невролога.
Генетический дефект хромосомы 19 является повреждением мышечных клеток, которые нельзя лечить по сей день. Причина, вероятно, заключается в изменении мембранной системы мышечных клеток. Мистастическая дистрофия может прогрессировать до полного разрушения мышечных клеток. Во-первых, мышцы поражаются в лицо, руки, предплечья, нижние ноги и ноги. Болезнь может возникать у мужчин и женщин всех возрастов.
Нередко у детей развивается адипозогенитальный синдром, иногда и другие признаки эндокринной недостаточности. Часто находят изменения в костной системе: деформацию стоп, грудной клетки, позвоночника, диффузный остеопороз.
Отличительной особенностью формы Дюшенна является высокая степень гиперферментемии уже на ранних стадиях развития процесса. Так, уровень специфического для мышечной ткани фермента - креатининфосфокиназы - в сыворотке крови может превышать в десятки и даже сотни раз нормальные показатели. Резкое (в 10-100 раз) увеличение креатининфосфокиназы (КФК) при нервно-мышечной патологии должно побуждать к обсуждению прежде всего следующих заболеваний: болезни Дюшенна, болезни Беккера, полиомиозита и дерматомиозита, пароксизмалыюй миоглобулинурии, дистальной миодистрофии. Лишь в далеко зашедших стадиях болезни степень гиперферментемии постепенно снижается. Имеются сообщения о повышении КФК на стадии внутриутробного развития.
Типичным является отсроченная релаксация мышц после мышечного напряжения. Последствиями являются слабость мышц и ограничения движения в ногах, руках и руках, ухудшение тонкой моторики. Некоторые больные мышцы особенно сильны, потому что типичный тканевый состав подстилающих мышечных волокон встроен в жир и соединительную ткань.
Он также упоминается как спинной мозг, влияющий на мышечный спазм. Существует до 30 различных форм спинальной мышечной атрофии. Наиболее частой формой является проксимальная спинальная мышечная атрофия, описанная здесь. Он обозначается следующим образом после начала фюзеляжа.
Миодистрофия Дюшенна передается по рецессивному, сцепленному с Х-хромосомой типу. Ген локализован в коротком плече Х-хромосомы. Довольно высока частота мутации гена (30%), что объясняет большое количество спорадических случаев.
Мутация (чаще всего делеция) приводит к половому или почти полному отсутствию продукта гена - структурного белка дистрофика. Физиологическая роль дистрофика до конца не установлена. Он обнаруживается в больших концентрациях в области сарколеммы, играя, видимо, определенную роль в поддержании целости этой мембраны. Отсутствие дистрофика вызывает структурные изменения в сарколемме, что в свою очередь приводит к потере внутриклеточных компонентов и повышенному входу кальция, что в конечном счете ведет к гибели миофибрилл. Полагают, что дефицит дистрофика в синаптических зонах корковых нейронов является причиной задержки психического развития.
Причиной, вероятно, является генетический дефект. Нервные клетки в спинном мозге, передние роговые клетки инфицированы. Предположительно, причиной является нервная система нервов. Это касается только двигательной нервной системы. Части нервной системы, которые ответственны за ощущение прикосновения, восприятие боли и температуры, остаются нетронутыми. Функция мочевого пузыря и прямой кишки не ухудшается.
Обозначение также представляет собой потерю мышц, обусловленных нервами. Причиной почти всегда является генетический дефект, оболочки нервных волокон становятся аномально толстыми, или сами нервные оболочки сами разрушаются. На нервные волокна на руках и ногах влияет.
Для медико-генетического консультирования очень важно установление гетерозиготного носительства. При миодистрофии Дюшенна у гетерозигот приблизительно в 70% случаев выявляются субклинические, а иногда и явные признаки мышечной патологии - некоторое уплотнение и даже увеличение икроножных мышц, быстрая утомляемость мышц при физической нагрузке, изменения на ЭМГ и при патоморфологическом исследовании мышечных биоптатов. Наиболее часто у гетерозиготных носительниц выявляется увеличение активности креатининфосфокиназы.
Интенсивность скорости нервной проводимости замедляется. Начало в нижних конечностях с мышечной потерей и сопутствующей мышечной слабостью. Симптомы затем поднимаются на нижних конечностях, позже воздействуя на руки и предплечья. Сенсорные нарушения низки. возможно Возможны вегетативные нарушения, такие как слишком много или слишком мало образования пота и нарушения основного кровотока. Могут возникнуть небольшие спастические симптомы на ногах.
Мышечные слабости возникают в результате мышечной слабости, пока мышцы полностью не сработают, что приводит к значительным серьезным ограничениям движения на ногах, руках и руках. Мышцы - очень важная часть нашего тела. Без мышц тело теряет способность двигаться и выполнять различные действия. На самом деле, без человеческой мышечной системы, вы, вероятно, не сможете выжить. Это связано с тем, что большинство органов в пищеварительной системе состоят из мышц и даже самого сердца, которое накачивает кровь, также является мышцей.
При наличии клинической картины миодистрофии Дюшенна у лиц женского пола следует в первую очередь исключить возможность аномалии по Х-хромосоме - синдром Шерешевского-Тернера (ХО), синдром Морриса (XY) или мозаицизм по этим синдромам.
Мышечная дистрофия Дюшенна, начинающая развиваться еще в пренатальном периоде, является по сути врожденной миопатией и может быть диагностирована вскоре после рождения путем проведения мышечной биопсии и определения активности КФК.
Миодистрофия Беккера. Наряду с тяжелой, злокачественной формой Х-сцепленной миодистрофией Дюшенна существует доброкачественная форма - болезнь Беккера. По клиническим симптомам она очень напоминает форму Дюшенна, однако начинается, как правило, позднее - в 10-15 лет, течет мягко, больные длительно сохраняют работоспособность, в возрасте 20- 30 лет и позже еще могут ходить. Фертильность не снижена, поэтому заболевание иногда прослеживается в нескольких поколениях семьи: больной мужчина через свою дочь передает заболевание внуку (“эффект деда”). Начальные симптомы, как и при болезни Дюшенна, проявляются слабостью в мышцах тазового пояса, затем в проксимальных отделах нижних конечностей. У больных изменяется походка, они испытывают затруднение при подъеме по лестнице, при вставании с низкого сиденья. Характерны псевдогипертрофии икроножных мышц. Ретракция пяточных (ахилловых) сухожилий выражена менее резко, чем при болезни Дюшенна.
При этой форме не отмечается нарушений интеллекта, кардиомиопатия отсутствует или выражена незначительно.
Как и при других Х-сцепленных миодистрофиях, при форме Беккера значительно повышается активность КФК, хотя и в меньшей степени, чем при болезни Дюшенна, не превышая 5000 ед. Ген болезни Беккера, как и болезни Дюшенна, локализуется в коротком плече Х-хромосомы; вероятно, оба локуса тесно связаны между собой или являются аллельными. В отличие от болезни Дюшенна, при которой дистрофии практически отсутствует, при болезни Беккера синтезируется аномальный дистрофии. Отличия обнаруживаются и при мышечной биопсии. При миодистрофии Беккера мышечные волокна обычно неокруглы, гиалиновые волокна, характерные для миодистрофии Дюшенна, наблюдаются крайне редко.
Миодистрофия Ландузи-Дежерина (лицелопаточно-плечевая миодистрофия). Заболевание передается по аутосомно-доминантному типу с высокой пенетрантностью, но несколько вариабельной экспрессивностью. Встречается гораздо реже, чем миодистрофия Дюшенна (0,4 на 100 тыс. населения). Предполагают, что ген этого заболевания локализован в 4-й хромосоме. Женщины болеют чаще мужчин (3:1), Физические перегрузки, интенсивные занятия спортом, а также нерационально проводимая лечебная физкультура могут способствовать более тяжелому течению болезни.
Миодистрофия Ландузи-Дежерина - сравнительно благоприятно текущая форма мышечной патологии. Начинается она еще в возрасте около 20 лет, иногда позже. Однако в семейных случаях заболевания, когда можно проследить за младшими членами семьи в динамике, удается выявить некоторую слабость мышц, например мышц лица, и в более раннем возрасте.
Мышечная слабость и атрофия вначале появляются в мышцах лица или плечевого пояса. Постепенно эти нарушения распространяются на мышцы проксимальных отделов рук, а затеми на нижние конечности. В большинстве случаев вначале поражаются мышцы передней поверхности голеней (с развитием свисающей стопы), затем мышцы проксимальных отделов ног. На высоте заболевания грубо страдают круговые мышцы глаза и рта, большая грудная, передняя зубчатая и нижние отделы трапециевидной мышцы, широчайшая мышца спины, двуглавая, трехглавая мышцы плеча. Характерен внешний вид больных: типичное лицо миопата с “поперечной улыбкой” (“улыбка Джоконды”), протрузией верхней губы (“губы тапира”), резко выраженные крыловидные лопатки, своеобразная деформация грудной клетки с уплощением ее в переднезаднем направлении и ротацией внутрь плечевых суставов. Нередко имеется асимметрия поражения, даже в пределах одной мышцы (например, круговой мышцы рта). Может наблюдаться псевдогипертрофия икроножных, дельтовидных мышц, иногда мышц лица. Контрактуры и ретракции выражены умеренно. Сухожильные рефлексы длительное время бывают сохранены, но иногда снижаются уже на ранней стадии.
Признаки поражения сердечной мышцы выявляются редко. Активность сывороточных ферментов увеличена незначительно и может быть нормальной. Интеллект не страдает. Продолжительность жизни в большинстве случаев не снижается. Представляет интерес тот факт, что ЭМГ при миодистрофии Ландузи-Дежерина нередко не совсем типична для мышечного уровня поражения. У некоторых больных (членов одной семьи) может наблюдаться снижение амплитуды биопотенциалов, интерференционный тип кривой, у других, наоборот, уменьшение частоты и гиперсинхронная активность, иногда с типичным ритмом частокола. Следует помнить о спинальном варианте, имитирующем болезнь Ландузи-Дежерина.
Миодистрофия Эрба-Рота (конечностно-поясная миодистрофия). Передается по аутосомно-рецессивному типу, оба пола страдают одинаково. Начало заболевания в большинстве случаев относится к середине 2-го десятилетия жизни (14-16 лет), однако описана как ранняя, псевдодюшенновская форма, когда первые симптомы проявляются в возрасте до 10 лет и заболевание протекает тяжело, так и поздний вариант с началом после 30 лет.
Течение заболевания может быть быстрым или более медленным, в среднем полная инвалидизация наступает через 15-20 лет от начала появления первых симптомов. Миодистрофия начинается либо с поражения мышц тазового пояса и проксимальных отделов ног (форма Лейдена-Мебиуса), либо с плечевого пояса (форма Эрба). В некоторых случаях плечевой и тазовый пояса поражаются одновременно. Довольно значительно страдают мышцы спины и живота. У больных имеется характерная “утиная” походка, затруднено вставание из положения лежа и сидя, подчеркнут поясничный лордоз. Мышцы лица в большинт стве случаев не страдают. Для этой формы малохарактерны контрактуры и псевдогипертрофии. Могут иметь место концевые атрофии и сухожильные ретракции. Интеллект обычно сохранен. Сердечная мышца большей частью не поражена. Уровень ферментов в сыворотке крови, как правило, повышен, однако не столь резко, как при Х-сцепленной миодистрофии. Есть указания, что у больных мужского пола уровень КФК выше, чем у больных женщин. Имеется значительная разница в экспрессивности мутантного гена у разных членов семьи - наряду с тяжелой клинической картиной могут быть сравнительно легкие и даже стертые клинические симптомы. Смерть обычно наступает от легочных осложнений.
Поскольку клинику конечностно-поясной миодистрофии особенно охотно имитируют нервно-мышечные заболевания иного характера, необходимо, особенно в спорадических случаях и при позднем начале заболевания, проводить тщательное клиническое обследование для исключения спинальной амиотрофии, полимиозита, метаболических, эндокринных, токсических, лекарственных, карциноматозных миопатий. В прошлом имела место явная гипердиагностика этой формы мышечных дистрофий.
Лечение мышечных дистрофий. Терапевтические возможности при мышечных дистрофиях весьма ограничены. Этиологического и патогенетического лечения практически не существует. Симптоматическое лечение направлено прежде всего на предотвращение развития контрактур, поддержание имеющейся мышечной силы и, возможно, на некоторое снижение скорости развития атрофии. Основная задача состоит в том, чтобы максимально продлить период, в течение которого больной способен самостоятельно передвигаться, так как в лежачем положении быстро нарастают контрактуры, сколиоз, дыхательные расстройства. Лечебный комплекс должен включать в себя лечебную гимнастику, массаж, ортопедические мероприятия, медикаментозную терапию.
Лечебная гимнастика состоит из пассивных и активных движений, выполняемых во всех суставах в различных положениях: стоя, сидя, лежа, при различном положении конечностей. Активные движения предпочтительнее выполнять в изометрическом режиме. Занятия гимнастикой необходимо проводить регулярно по нескольку раз в день. В то же время следует предостеречь от чрезмерных упражнений, особенно сопровождающихся перерастяжением мышц. Важное значение (особенно после иммобилизации больного) имеют дыхательные упражнения.
Ортопедические мероприятия консервативного (специальные шины) и оперативного характера (ахиллотомия, пересечение икроножной мышцы), направленные на коррекцию контрактур и формирующихся патологических установок конечностей, также имеют целью сохранить возможность самостоятельного передвижения. При этом в каждом случае необходимо индивидуально взвесить предполагаемую пользу и возможный вред от оперативного вмешательства. Следует учитывать, что нередко (в частности, при выраженном гиперлордозе и слабости четырехглавой мышцы бедра) эквиноварусная установка стоп имеет компенсаторное значение и после проведения, например, ахиллотомии больной может оказаться окончательно обездвиженным. При развивающихся контрактурах рекомендуется проводить осторожное растяжение мышц до 20-30 раз в день с последующим наложением шины на время сна.
Медикаментозная терапия предполагает назначение препаратов метаболического действия, направленных на восполнение энергетического и белкового дефицита, однако их эффективность весьма сомнительна. Применяют антагонисты кальция (в связи с выявленным при болезни Дюшенна дефектом клеточных мембран, приводящим к повышенному поступлению кальция внутрь клетки), иммуномодуляторы, фосфорсодержащие соединения (АТФ, фосфаден), витамин Е (100мг внутрь 3 раза в день). Показано, что при болезни Дюшенна применение преднизолона (0,75 мг/кг в сутки) может драматически увеличивать силу мышц, однако этот эффект сохраняется неболее года и в целом не влияет на исход заболевания. В связи с серьезными побочными эффектами, возникающими ври длительном применении препарата, его использование нецелесообразно. Оценки эффекта анаболических стероидов противоречивы и их назначение зачастую сопряжено с неоправданным риском. Оценивая эффект тех или иных препаратов при болезни Дюшенна, следует учитывать, что при умеренной выраженности заболевания у больных в возрасте 3-6 лет может отмечаться относительная стабилизация состояния, связанная с возрастным развитием мышечной системы, приобретением двигательных навыков, что может в какой-то степени временно скомпенсировать непрерывно текущий дистрофический процесс.
Определенное значение имеет коррекция питания больного, рекомендуется диета с высоким содержанием белка и низким содержанием жиров и пониженной калорийностью при оптимальном содержании витаминов и микроэлементов. Важную роль играет психологическая поддержка больного, продолжение обучения, правильная профессиональная ориентация.
Страница 44 из 44
Скелетные мышцы вовлекаются в патологический процесс при разнообразных дегенеративных, обменных и воспалительных заболеваниях. В большинстве случаев при этом происходит дегенерация мышечных волокон, а при хронических формах - их замещение соединительной тканью и жиром. Проксимальные группы мышц повреждаются более значительно, чем дистальные, как и нижние конечности по отношению к верхним. Больной ребенок отличается так называемой утиной (переваливающейся) походкой, не способен бегать, подниматься по лестнице и вставать, если находится в положении сидя. Сухожильные рефлексы у него угнетены, степень их угасания пропорциональна степени ослабления мышечной силы. Чувствительность не нарушается.
К диагностически ценным лабораторным методам относится определение активности ферментов, особенно креатинфосфокиназы, в сыворотке. Этот фермент, катализирующий реакцию: фосфокреатин + АДФ-креатин + АТФ, присутствует главным образом в клетках головного мозга и мышечной ткани. При некоторых диффузных мышечных заболеваниях, особенно при мышечной дистрофии, его избыточные количества проникают в межклеточное пространство и кровь. У больных обычно повышена активность сывороточной лактатдегидрогеназы и глутаминощавелевоуксусной трансаминазы, однако широкое распределение их в других тканях, включая печеночную, уменьшает специфичность теста. Обычно для уточнения диагноза требуется биопсия мышечной ткани.
Воспалительные болезни мышц. Воспаление мышечной ткани сопровождает некоторые инфекции, особенно трихинеллез, токсоплазмоз и вызванные вирусом Коксаки. Нередко оно бывает компонентом коллагеновых болезней, в том числе дерматомиозита, красной волчанки, узелкового периартериита и ревматоидного артрита.
Полимиозит. Диффузное изолированное воспаление мышц неизвестной этиологии называют полимиозитом. Для него типичны быстрое прогрессирующее течение, слабость и боль в проксимальных группах мышц. Часто в процесс вовлекаются мышцы шеи, в связи с чем ребенку становится трудно поднять голову и удерживать ее в этом положении. К лабораторным признакам воспаления мышц относится увеличение СОЭ и числа лейкоцитов. Однако их отсутствие не исключает полимиозита. Уровень сывороточных ферментов обычно повышен. В мышечном биоптате определяют дегенерацию и частичную регенерацию волокон и их инфильтрацию лимфоидными клетками. Дифференцировать полимиозит от мышечной дистрофии и дерматомиозита трудно. Он может представлять собой атипичную форму дерматомиозита, хотя гистологическая картина при этих двух состояниях несколько различна: для дерматомиозита характерен васкулит, обычно отсутствующий при полимиозите. Прогноз при последнем несколько благоприятнее. Лечение кортикостероидами сопровождается эффектом, но при их отмене может наступить рецидив.
Прогрессирующий оссифицирующий миозит. Этиология этого редко встречающегося заболевания соединительной ткани и мышц неизвестна. Сообщается, что им страдают родные братья и сестры, в том числе близнецы, и передается оно кровным родственникам по прямой линии. Предполагают, что наследуется болезнь по аутосомно-доминантному типу. Мальчики заболевают в 2-3 раза чаще девочек.
Патологические признаки зависят от стадии заболевания. На ранних стадиях местные отеки и воспалительные клеточные инфильтраты находят в мышцах и сухожилиях. Позднее участки воспаления замещаются грануляционной тканью и в конце концов в очагах поражения формируются участки хрящевой и костной ткани.
Почти у 75% больных детей выявляют врожденные пороки развития, чаще всего недоразвитие пальцев и анкилоз фаланг I пальцев ног и недоразвитие I пальцев рук, полидактилию, искривление пальцев, синдактилию (ноги), деформацию ушных раковин, глухоту, отсутствие зубов. Те же врожденные пороки могут быть у родственников больного, у которых не развилось прогрессирующее заболевание соединительной ткани и мышц. Возраст, в котором может начаться оссифицирующий миозит, варьирует от момента рождения до старшего детского возраста. Обычно различают три стадии его: 1) на местах незначительных местных травм появляются ограниченные, часто теплые и мягкие на ощупь тестообразные припухлости мягких тканей; 2) через несколько дней симптомы воспаления исчезают, а очаг поражения затвердевает; 3) происходит окостенение пораженного участка. Периодически появляются новые очаги, преимущественно в областях шеи и спины. Первичным признаком может стать кривошея, если процесс развился в грудиноключично-сосцевидной мышце. В конце концов окостенение распространяется на многие сухожилия и связки. Наступает анкилоз позвоночника и суставов рук и ног (рис. 21-5). Воспаление может распространиться на височно-нижнечелюстные суставы, в связи с чем затрудняются жевательные движения. Костные выросты могут выступать через кожу. В юношеском возрасте заболевание часто приводит к полному обездвижению и смерти вследствие дыхательной недостаточности и прекращения дыхания, хотя есть сообщения о случаях выживания. При оссифицирующем миозите велика опасность развития остеогенной саркомы.
Рис. 21-5. Ребенок с прогрессирующим оссифицирующим миозитом (типичная поза с ригидностью мышц шеи и спины).
Иногда патологический процесс бывает ограничен местом предшествовавшей травмы мягких тканей (miositis ossificans circumscripta). Широко распространенная кальцификация мышечной ткани может произойти и при хроническом полимиозите и дерматомиозите.
Результаты лабораторных методов исследований не имеют диагностической ценности.
Уровни в сыворотке кальция, фосфора, щелочной фосфатазы, а также активность креатинфосфокиназы и других ферментов остаются в норме. Костная ткань в очаге повреждения не отличается по строению от нормы.
Существующие методы лечения неудовлетворительны. В некоторых случаях отмечено замедление развития заболевания при применении АКТГ и других кортикостероидов. Их роль в конечном результате лечения вызывает сомнения.
Эндокринные и обменные миопатии. Миопатия при гипертиреозе представляет собой довольно редкое осложнение. Для него типичны птоз, двусторонний парез лицевых мышц и мышц проксимальных отделов конечностей. При этом некоторые симптомы гипертиреоза могут маскироваться мышечной слабостью, однако налицо остаются тахикардия, усиленное потоотделение и увеличение щитовидной железы. Сухожильные рефлексы в отличие от многих других форм миопатии остаются в норме. После коррекции гипертиреоза мышечная слабость постепенно исчезает.
Миопатия при гипотиреозе. Гипотиреоз у грудных детей может быть связан с мышечными слабостью и гипотонией. У детей старшего детского возраста с микседемой замедляются сокращения и расслабления мышц, в некоторых случаях отмечается мышечная гипертрофия (синдром Дебре - Семелена). Совокупность таких признаков, как слабость и гипертрофия мышц, дает возможность предположить мышечную дистрофию.
Миопатия при лечении кортикостероидами. Она может осложнять болезнь Иценко - Кушинга, но чаще развивается при лечении большими дозами синтетических стероидов. Слабость особенно заметна в мышцах тазового пояса, что проявляется в переваливающейся (утиная) походке, затруднениях при подъеме по лестнице и попытке встать из положения сидя. Коленный рефлекс отсутствует. Может наступить истончение мышц. Миопатические изменения в мышечной ткани обычно незначительны даже при ее выраженной слабости. Мышечная сила после отмены кортикостероидов восстанавливается медленно (в течение нескольких месяцев).
Миопатия при гиперпаратиреозе. Гиперпаратиреоз может быть связан со слабостью и гипорефлексией, обусловленными гиперкалиемией. Обычно они быстро исчезают после паратиреоидэктомии.
Дефицит карнитина (липидная миопатия) сопровождает накопление в больших количествах липидов в мышцах и нарушение в связи с этим энергетического обеспечения последних. Карнитин относится к обязательным компонентам системы, обеспечивающей перенос жирных кислот с длинной цепью из цитозоля в митохондрии, где они подвергаются окислению. Мышечная слабость развивается при двух формах недостаточности карнитина.
Недостаточность карнитина в мышцах клинически представлена прогрессирующей слабостью их проксимальных групп, чаще у школьников и подростков. Иногда слабость интермиттирует и сочетается с миоглобинурией. При тяжелом течении может наступить паралич дыхательной мускулатуры. Уровень ферментов (креатинкиназа и альдолаза) в сыворотке повышается. На электромиограмме выявляют неспецифические изменения, свойственные миопатии. В биоптате мышц можно видеть большое количество капелек жира. Уровень карнитина в сыворотке не изменяется, но в мышцах снижается. Распознавание патологии имеет существенное значение, так как она может быть курабельной. Нередко ее принимают за мышечную дистрофию. Эффект может наступить после применения внутрь 100 мг/(кг\сут) карнитина. В некоторых случаях эффективно лечение кортикостероидами.
- Системная недостаточность карнитина проявляется прогрессирующей миопатией, в том числе кардиомиопатией, и дисфункцией печени, сопровождающейся клиникой печеночной энцефалопатии по типу синдрома Рея. От последнего карнитиновая недостаточность отличается рецидивирующим течением и сохраняющейся между периодами обострения энцефалопатии выраженной мышечной слабостью. Уровень креатинфосфокиназы в сыворотке заметно повышен, количество карнитина уменьшено как в сыворотке, так и в мышцах. Изменения в биоптате аналогичны таковым при дефиците карнитина в мышечной ткани. Сходные клинические и морфологические изменения, в том числе карнитиновую недостаточность, можно выявить при нарушении обмена органических кислот, например, при метилмалоновой и глутаровой ацидурии (вторичная карнитиновая недостаточность).
Рис. 21-6. Ребенок с врожденным отсутствием левой большой грудной мышцы.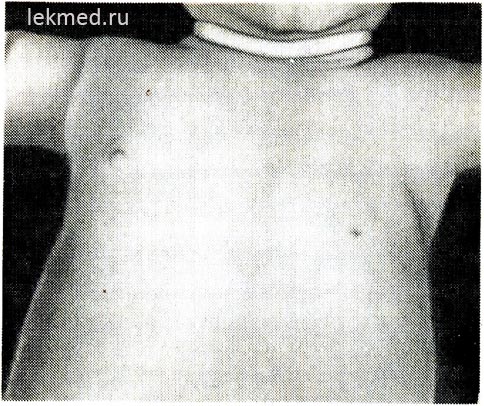
Обращают на себя внимание отсутствие передней подмышечной складки и низкорасположенный сосок.
Лечение состоит в соблюдении больным диеты, богатой углеводами и обезжиренной, и приеме карнитина в суточной дозе 100 мг/кг.
Врожденные мышечные дефекты. Врожденное отсутствие мышцы. Недоразвитие мышц может быть довольно распространенным и приводить к полной блокаде движений в суставах или врожденному артрогрипозу. Как врожденный дефект чаще всего отсутствует одна мышца. К довольно распространенной аномалии относится отсутствие стернальной части большой грудной мышцы (рис. 21-6), в некоторых случаях этот дефект сочетается с синдактилией на пораженной стороне (синдром Поланда). Отсутствие грудной мышцы часто сопровождает мышечную дистрофию. Врожденное отсутствие брюшных мышц живота часто связано с дефектами развития мочевых путей.
Рис. 21-7. Деформация шеи и асимметрия лица у мальчика с врожденной кривошеей, не леченного с 12-летнего возраста.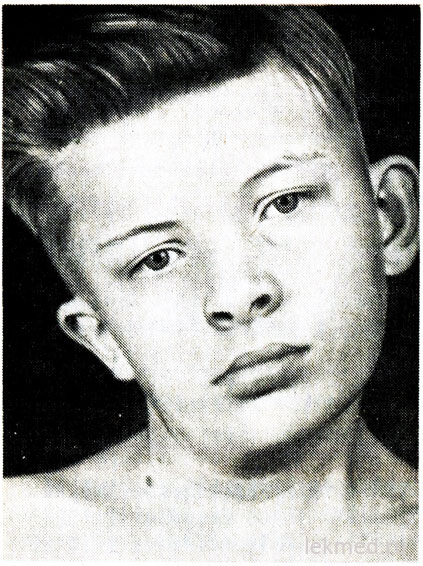
Врожденная кривошея обусловлена односторонним укорочением или контрактурой грудиноключично-сосцевидной мышцы. Голова больного наклонена в сторону контрактуры, а подбородок направлен книзу в противоположную сторону (рис. 21-7). При попытке коррекции положения головы ощущается значительное сопротивление мышц. В пораженной мышце прощупываются участки уплотнения. Причина дефекта неясна, в течение длительного времени его считали результатом родовой травмы. Однак кривошея встречается у детей, рожденных с помощью операции кесарева сечения; это дает возможность предположить, что в некоторых случаях причина дефекта относится к внутриутробному периоду. Кривошею следует дифференцировать от патологического наклона головы вследствие деформации шейных позвонков, например при аномалии Клиппеля - Вейля, и от переломов или вывихов шейных позвонков. Их исключают с помощью рентгенографического исследования. У детей старшего возраста наклон головы может быть при косоглазии, дистонии, опухолях задней черепной ямки и шейного отдела спинного мозга, оссифицирующем миозите, шейном лимфадените или диафрагмальной грыже. В большинстве случаев врожденная кривошея поддается коррекции с помощью лечебной гимнастики. Однако при хронической форме кривошея приводит к асимметричному развитию лица и головы (см. рис. 21-7), в связи с чем может потребоваться рассечение мышцы, что вызвано косметическими целями.
Врожденные миопатии. В эту группу входят несколько редких форм наследуемых заболеваний, при которых мышечные слабость и гипотония появляются с грудного возраста (см. табл. 22-1). Их точная диагностика имеет большое значение с точки зрения прогноза. В целом для нормальной жизнедеятельности и продолжительности жизни он благоприятен в отличие от болезни Верднига - Гоффманна или врожденной мышечной дистрофии. Выявлению врожденных миопатий обычно способствует биопсия мышц.
- Болезнь центрального стержня. Центральная часть мышечных волокон окрашена аномально, но однородно. При электронномикроскопическом исследовании выявляют уменьшение количества митохондрий и обеднение саркоплазматического ретикулума в центральной части волокон.
Немалиновая миопатия. Термин «немалиновая» объясняется тем, что в мышечных волокнах определяются нитеподобные структуры.
Рис. 21-8. Миотоническое сокращение языка (а) при резком ударе перкуссионным молоточком по его правой половине и век (б) у ребенка с гиперкалиемической формой семейного периодического паралича.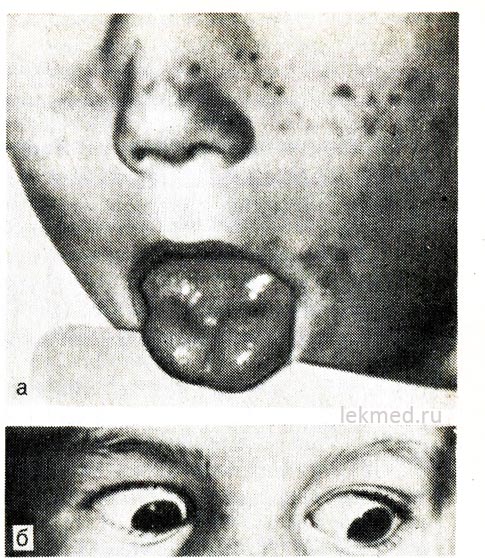
При взгляде вниз веко остается сокращенным.
Данные электронно-микроскопического исследования свидетельствуют о том, что это результат изменений Z-полосок миофибрилл.
Митохондриальные миопатии. Сообщается о некоторых формах миопатий, при которых наиболее важные изменения происходят в митохондриях мышечных волокон. Они могут заметно увеличиваться как в числе, так и в размерах. Мышечные слабость и гипотония могут определяться уже в грудном возрасте, но иногда заметно прогрессируют только в школьном. Кардиомиопатия, энцефалопатия и лактатацидемия часто сопровождают миопатии этой группы.
Миотония
. Это состояние служит признаком различных мышечных заболеваний, например дистрофической миотонии, гиперкалиемической формы семейного пароксизмального паралича и болезней накопления гликогена. Миотония определяется как значительное замедление расслабления мышц после их произвольных или вынужденных сокращений. Клинически она проявляется в неспособности разжать кулак или в видимом длительном сокращении мышц вслед за их стимуляцией, выражающейся в резком раздражении (рис. 21-8). Это можно наблюдать, если ударить перкуссионным молоточком по поверхностной группе мышц, например по мышцам языка или ладонной поверхности в области возвышения I пальца. Миотония подтверждается данными электромиографии. В этом случае заметна характерная спонтанная активность мышц после их расслабления или произвольного сокращения (миотонические разряды).
Врожденная миотония (болезньТомсена)
. Единственным признаком этого заболевания, наследуемого по доминантному типу, служит миотония. Оно может проявиться в грудном возрасте в виде замедления глотательных движений и рвоты вслед-
ствие неспособности к нормальному расслаблению мышц глотки. В более старшем детском возрасте миотония проявляется как неспособность больного разжать сжатые в кулак пальцы. При первой попытке осуществить какое-то движение мышцы ребенка становятся твердыми. При многократном повторении того же движения они несколько расслабляются. Так, например, больной ребенок испытывает большие затруднения в начале акта ходьбы. Первые несколько шагов он обычно осуществляет очень неуверенно и медленно. Через несколько секунд походка становится нормальной или почти нормальной. Симптомы миотонии усугубляются при неблагоприятном эмоциональном состоянии больного и охлаждении организма. Мышечная сила остается в норме, мышцы достаточно развиты и нередко заметно увеличены, что создает ложное впечатление атлетического сложения больного.
Диагноз основывается на клинических данных и данных электромиографии. Активность сывороточных ферментов находится в пределах нормы. Единственным гистологическим признаком служит гипертрофия мышечных волокон.
От дистрофической миотонии заболевание отличается отсутствием мышечных слабости и атрофии и дистрофических изменений в биоптате мышечной ткани. Лечение новокаином или сульфатом хинидина сопровождается эффектом и показано при функциональных нарушениях. Течение заболевания обычно доброкачественное, и состояние больного может с возрастом улучшаться.
Пароксизмальные параличи.
Для этой группы заболеваний характерна периодическая мышечная слабость с полным или почти полным восстановлением силы мышц в период между приступами. К ней относится и дефицит мышечной фосфорилазы (болезнь МакАрдла).
Гиперкалиемический пароксизмальный паралич.
Наследственная эпизодическая адинамия, или парамиотония, передается по доминантному типу и особенно тяжело протекает у лиц мужского пола. Начинается обычно в раннем детском (иногда в грудном) возрасте. Приступы возникают в период отдыха после тяжелой мышечной нагрузки. Быстро развивается слабость, которая может продолжаться в течение нескольких часов. Она особенно ощущается в ногах; функция дыхания обычно не нарушена. Часто адинамии сопутствует миотония, сохраняющаяся между приступами, что наиболее ярко проявляется в виде запаздывания движения век при взгляде вниз (см. рис. 21-8, б).
Во время приступа уровень калия в сыворотке часто повышен, однако для того, чтобы определить это с достоверностью, могут потребоваться многократные исследования во время нескольких приступов. Искусственно спровоцировать приступ можно с помощью калиевой нагрузки (2-3 г внутрь), однако ее следует проводить только под контролем ЭКГ. Повторные приступы купируют диакарбом. Тяжелые формы заболевания характеризуются развитием хронической, слабо выраженной слабости и дистрофическими изменениями в мышцах.
Гипокалиемический пароксизмальный паралич
. Семейный пароксизмальный паралич, также наследуемый по доминантному типу, особенно тяжело протекает у мальчиков. В отличие от гиперкалиемической формы первый приступ появляется в позднем детском или раннем подростковом возрасте. Причиной служит употребление обильной пищи, богатой углеводами, или отдых после физической нагрузки. Обычно приступ начинается на следующее утро после тяжелой физической нагрузки и обильного ужина. Он характеризуется мышечной слабостью и арефлексией. Может быть нарушена функция дыхания. Может присоединиться аритмия, включая желудочковые экстрасистолию и тахикардию. Приступы могут продолжаться более 24 ч. В паралитической фазе уровень калия в сыворотке обычно снижается (2-3 ммоль/л). Основной дефект неизвестен. У больных с повторными тяжелыми приступами развиваются хроническая мышечная слабость и патологические изменения в мышцах. Лечение во время приступов состоит в приеме калия хлорида; его начальная доза составляет 2-3 г. Диакарб способствует уменьшению частоты приступов.
Пароксизмальная миоглобинурия (идиопатическая миоглобинурия)
. Идиопатическая миоглобинурия представляет собой разнородную группу заболеваний, при которых приступы паралича с миоглобинурией возникают спонтанно или после интенсивной физической нагрузки. Заболевание наследуется по доминантному типу, сцеплено с Х-хромосомой. Мышцы, чаще всего икроножные и бедренные, во время приступа становятся болезненными и припухлыми. Моча приобретает темно-красный или коричневый цвет. Миоглобинурия может обусловить некроз почечных канальцев, что приводит к летальному исходу в результате почечной недостаточности.
Диагноз подтверждается выявлением в моче миоглобулина. Положительная бензидиновая проба при отсутствии в моче эритроцитов подтверждает присутствие в ней миоглобина, особенно если в сыворотке не определяется гемоглобин. Гемоглобин определяют с помощью спектрофотометрии. Пароксизмальную миоглобинурию следует отличать от болезни Мак-Ардла, недостаточности карнитинпальмитилтрансферазы и миоглобинурии после непривычной интенсивной физической нагрузки или травмы мышц у здорового человека. Миоглобинурия после тяжелой мышечной нагрузки встречается при псевдогипертрофической мышечной дистрофии (болезнь Дюшенна).
Лечение состоит в соблюдении постельного режима; при необходимости проводят искусственную вентиляцию легких. Для предотвращения почечной недостаточности необходимо назначать больному обильное питье.
Недостаточность карнитинпальмитилтрансферазы.
При дефиците этого фермента нарушается перенос длинноцепочечных жирных кислот в митохондриальные сегменты, в которых осуществляются окисление и продукция кетонов. Недостаточность изофермента II типа наследуется по рецессивному типу. Вследствие его дефицита нарушается кетогенез в тканях, в том числе в мышечной и печеночной. Первые признаки болезни появляются чаще у детей школьного и подросткового возрастов. Они заключаются в повторных эпизодах болей в мышцах, слабости и повышении температуры тела после физической нагрузки или голодания. Миоглобинурия, сопровождающая приступы, может привести к почечной недостаточности. Голодание приводит к гипогликемии. Между приступами дети выглядят здоровыми. Заболевание необходимо дифференцировать от других состояний, сопровождающихся периодическими слабостью и миоглобинурией. Дифференциально-диагностической ценностью обладает метод определения активности карнитинпальмитилтрансферазы. Она снижается в мышечной и печеночной тканях, лейкоцитах и культуре фибробластов. Соблюдение диеты, состоящей из продуктов, обогащенных углеводами и обезжиренных, способствует уменьшению числа приступов.
Мышечные дистрофии. Эти аномалии относятся к группе семейных заболеваний, сопровождающихся дегенерацией мышечных волокон. Классификация мышечных дистрофий основана на таких признаках, как время начала, скорость прогрессирования, распределение поражений по группам мышц и тип наследования.
Псевдогипертрофическая мышечная дистрофия
. Детская, или форма Дюшенна, представляет собой наиболее распространенную форму мышечной дистрофии; ее частота составляет 0,14 на 1000 детей. В классической форме она протекает только у мальчиков, причем наследование, сцепленное с Х-хромосомой, происходит примерно у 50% пробандов. В остальных случаях заболевание обусловлено новыми мутациями. Сообщается о редкой форме мышечной дистрофии, по клинике идентичной форме Дюшенна, но наследуемой по рецессивному типу с одинаковой частотой заболевания мальчиков и девочек. Достоверно диагностировать заболевание редко возможно у ребенка в возрасте до 3 лет. В анамнезе обычно есть указания на то, что у ребенка было замедленное развитие двигательных функций, он поздно начал сидеть, ходить и бегать, что, естественно, свидетельствует о более раннем начале заболевания. Переваливающаяся (утиная) походка, трудности при подъеме по лестнице, гипертрофия икроножных мышц относятся к обычным клиническим проявлениям. В некоторых случаях в процесс вовлекаются также другие мышцы, в частности дельтовидная, плечелучевая, мышцы языка.
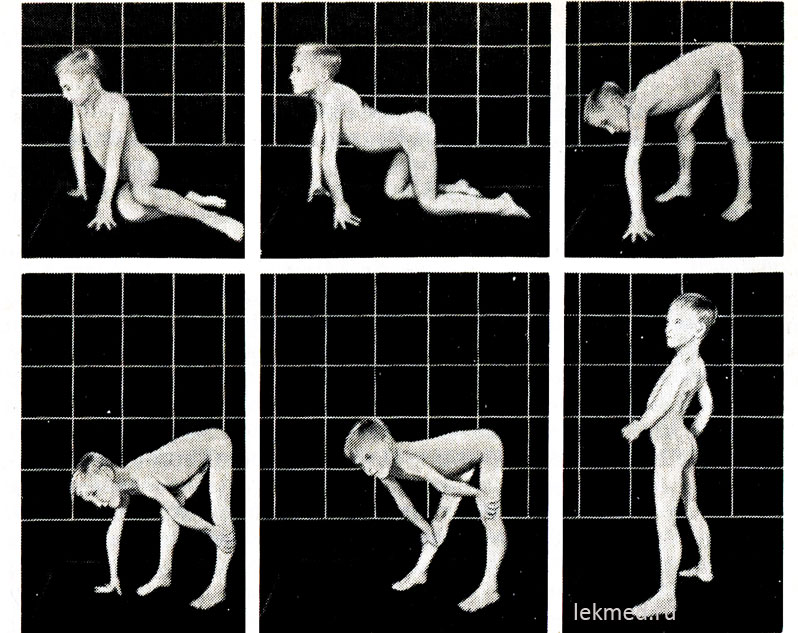
Рис. 21-9. Типичные позы, принимаемые при вставании с пола (симптом Говерса) ребенком в возрасте 7 лет с псевдогипертрофической миопатией.
В положении стоя (последняя фотография) отмечается значительно выраженный лордоз.
В начале заболевания гипертрофированные мышцы обладают значительной силой, но позднее она уменьшается (псевдогипертрофия), так как увеличение массы мышц происходит за счет их жировой инфильтрации. Сила гипертрофированной икроножной мышцы значительно превышает силу мышц передней поверхности ноги, что объясняет частые контрактуры пяточного сухожилия и хождение ребенка на пальцах. Слабость мышц тазового пояса выражается в характерной утиной (лордическая) походке и затруднениях, которые ребенок испытывает, когда встает из положения сидя на полу. При достаточно тяжелых формах мышечной дистрофии у ребенка отмечается симптом Говерса: вставая с пола, он прежде всего становится на колени, опираясь на руки, а затем поднимается, последовательно отталкиваясь руками от голеней, коленных суставов и бедер (рис. 21-9). Определить слабость мышц плечевого пояса можно, удерживая ребенка в приподнятом положении за подмышки. В норме он старается удержаться, прижимая руки к туловищу; при мышечной дистрофии он как бы проскальзывает через руки обследующего. Больной ребенок часто не может поднять руки над головой. В поздние стадии заболевания развивается значительная мышечная атрофия. Обычно к возрасту 12 лет ребенок уже не может ходить. Больные в 75% случаев умирают в возрасте до 20 лет. У большинства из них отмечается кардиомиопатия, которая в некоторых случаях служит причиной внезапной смерти. Если наследование сцеплено с Х-хромосомой, а заболевание началось в старшем детском возрасте, продолжительность жизни остается большой (мышечная дистрофия Беккера). Средний коэффициент умственного развития у детей с формой Дюшенна составляет 80; у 25% детей отмечается отставание умственного развития.
При дифференциальной диагностике мышечной дистрофии Дюшенна следует иметь в виду болезнь Верднига-Гоффманна у детей старшего грудного возраста и такие мышечные заболевания, как эндокринные миопатии, недостаточность карнитина, болезни накопления гликогена и полимиозит. Иногда при контрактурах пяточного сухожилия и хождении ребенка на пальцах ног можно предположить церебральный паралич, однако при мышечной дистрофии отсутствуют характерные для церебрального паралича спастичность и гиперрефлексия.
Диагноз основан на определении активности сывороточных ферментов, данных электромиографии и биопсии мышечной ткани. Активность ферментов, особенно креатинфосфокиназы, еще до развития клинических симптомов часто превышает нормы в 10 раз даже у детей грудного возраста. На электромиограмме выявляют прежде всего уменьшение продолжительности и снижение амплитуды двигательных потенциалов. Гистологические изменения состоят в дегенерации мышечных волокон. Они часто различаются по размеру и частично замещены жиром и соединительной тканью. Размер их ядер также варьирует. Диагноз может быть установлен при рождении путем определения активности креатинфосфокиназы. Методы выявления женщин-носителей еще не разработаны, несмотря на то что у 60-80% из них выявляют незначительное или умеренное повышение ее уровня. Эти признаки типичны более для детского возраста, нежели последующих периодов жизни.
Эффективных методов лечения не существует. Следует как можно больше способствовать поддержанию активного состояния больного и его способности ходить. Необходимо следить за тем, чтобы ребенок избегал интенсивной физической нагрузки, так как она может быть причиной разрыва мышечных волокон. В некоторых случаях хирургическое удлинение пяточного сухожилия способствует возможности ходить, однако продолжительный постельный режим после ортопедической коррекции может усилить мышечную атрофию. Важную роль играет генетическое консультирование.
Врожденная мышечная дистрофия. Заболевание наследуется по аутосомно-рецессивному типу и характеризуется мышечными гипотонией и слабостью у ребенка грудного возраста. Оно входит в группу состояний, определяемых как «вялый ребенок» (см. табл. 21-1). Начало заболевания относится к внутриутробному периоду. Иногда у новорожденного отмечают выраженную атрофию мышц, их контрактуры, ограниченную подвижность суставов. Дифференцирование от болезни Верднига- Гоффманна затруднено. Фасцикуляции языка, характерные для последней, отсутствуют при мышечной дистрофии. Сухожильные рефлексы угнетены, но полностью не утрачены. В процесс вовлекаются мышцы, участвующие в дыхании, включая диафрагму. При тяжелом течении смерть наступает в возрасте до 1 года вследствие дыхательной недостаточности; при более легких формах нормальная жизнеспособность сохраняется в течение длительного времени. Повышения активности сывороточных ферментов не отмечают, хотя в мышцах происходят дистрофические изменения.
Плечелопаточно-лицевая форма мышечной дистрофии. Эта достаточно легкая форма мышечной дистрофии наследуется по аутосомно-доминантному типу. Начинается она обычно в возрасте 10-20 лет и характеризуется слабостью и атрофией мышц лица и плечевого пояса. Лицо полностью амимично, больной не может закрыть глаза и произвести свист. Заболевание прогрессирует медленно и совместимо с нормальной продолжительностью жизни. Диагноз основан на клинических данных и типе наследования. Результаты биопсии мышечной ткани свидетельствуют о дистрофических изменениях в ней. Уровень креатинфосфокиназы в сыворотке может оставаться в пределах нормы или слегка повышается.
Тазовая форма мышечной дистрофии. Эта группа неоднородных нарушений характеризуется медленным прогрессированием мышечной дистрофии и наследуется по аутосомно-рецессивному типу. Начало заболевания относится к старшему детскому, подростковому или взрослому возрасту. Обычно поражаются мышцы тазового пояса.
Глазная форма миопатии. Дистрофические изменения происходят в основном в наружных глазных мышцах. Начинается заболевание в детском или подростковом возрасте. При нем прогрессируют птоз и ограничение движений глазных яблок. Иногда слабость распространяется на мышцы лица и шеи. Заболевание следует дифференцировать от миастении и параличей черепных нервов при опухолях ствола мозга.
Прогрессирующая офтальмоплегия, начинающаяся в детском или подростковом возрасте, связана с атипичной пигментной дегенерацией сетчатки и блокадой сердца (синдром Кернса - Сейерса). Обычно с ней связаны и прогрессирующая атаксия, отставание роста и полового созревания. Под сарколеммой мышц определяют большие скопления атипичных митохондрий. Генетическая природа этого процесса не установлена. Можно контролировать возможность внезапной смерти от нарушения сердечной проводимости с помощью кардиостимулятора.
Миотоническая дистрофия. Несмотря на то что миотоническая дистрофия начинается как будто у взрослого человека, ее начало все чаще регистрируют у детей грудного и более позднего детского возраста. Она наследуется по аутосомно-доминантному типу. Ее начало в детском возрасте свидетельствует о том, что миотонией страдает мать. В соответствии с этим внутриутробные факторы могут влиять на выраженность заболевания у ребенка. Уже в момент рождения у него может определяться мышечная гипотония, у него отсутствует способность сосать. Отставание физического и умственного развития обычно выявляется позднее. В раннем детском возрасте мышечные слабость и атрофия распространяются в основном на лицевые, челюстные и височные мышцы. Обычно отмечается двусторонний птоз. К диагностически значимым методам относятся перкуссия мышц, электромиография; типична для этих больных неспособность разжать сжатую в кулак кисть (см. Врожденная миотония). Слабость и атрофию мышц конечностей и тазового пояса (обычно дистальные группы) выявляют в старшем детском или подростковом возрасте. У взрослых этому заболеванию сопутствуют катаракта, облысение, атрофия яичек.
Диагноз основан на выявлении признаков миотонии, характерном распределении мышечной слабости, наследовании по доминантному типу, дистрофических изменениях мышц. В детском возрасте течение заболевания может быть неблагоприятным, часто ему сопутствует умственная отсталость. К юношескому возрасту мышечная слабость выступает на передний план. При функциональных нарушениях показано лечение новокаином и хинидином.

