L'aldosterone nell'uomo è il principale rappresentante degli ormoni mineralcorticoidi, derivati del colesterolo.
Sintesi
Viene effettuato nella zona glomerulosa della corteccia surrenale. Il progesterone, formato dal colesterolo, subisce un'ossidazione sequenziale nel suo percorso verso l'aldosterone. 21-idrossilasi, 11-idrossilasi e 18-idrossilasi. Alla fine si forma l’aldosterone.

Schema di sintesi degli ormoni steroidei (schema completo)
Regolazione della sintesi e della secrezione
Attivare:
- angiotensina II, rilasciato all'attivazione del sistema renina-angiotensina,
- maggiore concentrazione ioni potassio nel sangue (associato alla depolarizzazione della membrana, all'apertura dei canali del calcio e all'attivazione dell'adenilato ciclasi).
Attivazione del sistema renina-angiotensina
- Per attivare questo sistema ci sono due punti di partenza:
- diminuzione della pressione nelle arteriole afferenti dei reni, che viene determinato barocettori cellule dell'apparato iuxtaglomerulare. La ragione di ciò potrebbe essere qualsiasi violazione del flusso sanguigno renale - aterosclerosi arterie renali, aumento della viscosità del sangue, disidratazione, perdita di sangue, ecc.
- diminuzione della concentrazione di ioni Na+ nell'urina primaria nei tubuli distali dei reni, che è determinata dagli osmocettori delle cellule dell'apparato iuxtaglomerulare. Si verifica a seguito di una dieta priva di sale, con l'uso a lungo termine di diuretici.
Costante e indipendente dal flusso sanguigno renale, la secrezione di renina (basale) è mantenuta dal sistema nervoso simpatico.
- Quando si esegue uno o entrambi i punti della cella apparato iuxtaglomerulare vengono attivati e da essi l'enzima viene secreto nel plasma sanguigno renina.
- Per la renina nel plasma esiste un substrato: la proteina della frazione α2-globulina angiotensinogeno. Come risultato della proteolisi, un decapeptide chiamato angiotensina I. Successivamente, l'angiotensina I con la partecipazione Enzima di conversione dell'angiotensina(APF) diventa angiotensina II.
- I principali bersagli dell'angiotensina II sono i miociti lisci vasi sanguigni E corteccia della zona glomerulare ghiandole surrenali:
- la stimolazione dei vasi sanguigni provoca il loro spasmo e il ripristino pressione sanguigna.
- secreto dalle ghiandole surrenali dopo la stimolazione aldosterone, agendo sui tubuli distali dei reni.
Quando l'aldosterone agisce sui tubuli renali, il riassorbimento aumenta Ioni Na+, segue il sodio acqua. Di conseguenza, la pressione nel sistema circolatorio viene ripristinata e la concentrazione di ioni sodio aumenta nel plasma sanguigno e, quindi, nell'urina primaria, riducendo l'attività del RAAS.

Attivazione del sistema renina-angiotensina-aldosterone
Meccanismo di azione
Citosolico.
Obiettivi ed effetti
Colpisce ghiandole salivari, sui tubuli distali e sui dotti collettori dei reni. Rafforza nei reni riassorbimento degli ioni sodio e perdita di ioni potassio attraverso i seguenti effetti:
- aumenta la quantità di Na + ,K + -ATPasi di membrana basale cellule epiteliali,
- stimola la sintesi delle proteine mitocondriali e un aumento della quantità di energia generata nella cellula per il lavoro di Na + ,K + -ATPasi,
- stimola la formazione di canali per il Na sulla membrana apicale delle cellule epiteliali renali.
Patologia
Iperfunzione
La sindrome di Conn (aldosteronismo primario) – si verifica con gli adenomi della zona glomerulosa. È caratterizzata da una triade di sintomi: ipertensione, ipernatriemia, alcalosi.
Secondario iperaldosteronismo - iperplasia e iperfunzione delle cellule iuxtaglomerulari ed eccessiva secrezione di renina e angiotensina II. C'è un aumento della pressione sanguigna e la comparsa di edema.
Il sistema renina-angiotensina-aldosterone è un complesso di enzimi e ormoni che mantengono l'omeostasi. Regola l'equilibrio di sale e acqua nel corpo e i livelli di pressione sanguigna.
Meccanismo di funzionamento
La fisiologia del sistema renina-angiotensina-aldosterone ha origine al confine della corteccia e dove sono presenti le cellule iuxtaglomerulari che producono peptidasi (enzima) - renina.
La renina è un ormone e il collegamento iniziale del RAAS.
Situazioni in cui la renina viene rilasciata nel sangue
Esistono diverse condizioni in cui l'ormone entra nel flusso sanguigno:
- Ridotto flusso sanguigno nel tessuto renale - durante i processi infiammatori (glomerulonefrite, ecc.), con nefropatia diabetica, tumori renali.
- Diminuito (con sanguinamento, vomito ripetuto, diarrea, ustioni).
- Caduta dei livelli di pressione sanguigna. Le arterie dei reni contengono barocettori che rispondono ai cambiamenti della pressione sistemica.
- Variazione della concentrazione degli ioni sodio. Nel corpo umano ci sono gruppi di cellule che rispondono ai cambiamenti nella composizione ionica del sangue stimolando la produzione di renina. Il sale si perde quando sudorazione profusa, così come con il vomito.
- Stress, stress psico-emotivo. I reni sono innervati da nervi simpatici, che vengono attivati da influssi psicologici negativi.
Nel sangue, la renina incontra una proteina, l'angiotensinogeno, che viene prodotta dalle cellule del fegato e ne prende un frammento. Si forma l'angiotensina I, che è una fonte di azione per l'enzima di conversione dell'angiotensina (ACE). Il risultato è l'angiotensina II, che funge da secondo anello ed è un potente vasocostrittore sistema arterioso(costringe i vasi sanguigni).
Effetti dell'angiotensina II
Obiettivo: aumentare pressione arteriosa.
- Promuove la sintesi dell'aldosterone nella zona glomerulosa della corteccia surrenale.
- Colpisce il centro della fame e della sete nel cervello, provocando un appetito “salato”. Il comportamento umano viene motivato alla ricerca di acqua e cibo salato.
- Colpisce nervi simpatici, favorendo il rilascio di norepinefrina, anch'essa vasocostrittrice, ma meno debole nell'azione.
- Colpisce i vasi sanguigni, causandone lo spasmo.
- Partecipa allo sviluppo dell'insufficienza cardiaca cronica: favorisce la proliferazione, la fibrosi dei vasi sanguigni e del miocardio.
- Riduce
- Inibisce la produzione di bradichinina.

L'aldosterone è il terzo componente che agisce sui tubuli terminali dei reni e favorisce il rilascio di ioni potassio e magnesio dal corpo e il riassorbimento (riassorbimento) di sodio, cloro e acqua. A causa di ciò, il volume del fluido circolante aumenta, la pressione sanguigna aumenta e il flusso sanguigno renale aumenta. I recettori dell'aldosterone si trovano non solo nei reni, ma anche nel cuore e nei vasi sanguigni.
Quando il corpo raggiunge l'omeostasi, iniziano a produrre vasodilatatori (sostanze che dilatano i vasi sanguigni) - bradichinina e callidina. E i componenti del RAAS vengono distrutti nel fegato.
Schema del sistema renina-angiotensina-aldosterone

Come ogni sistema, il RAAS può fallire. La fisiopatologia del sistema renina-angiotensina-aldosterone si manifesta nelle seguenti condizioni:
- Danni alla corteccia surrenale (infezione, emorragia e trauma). Si sviluppa uno stato di carenza di aldosterone e il corpo inizia a perdere sodio, cloro e acqua, il che porta ad una diminuzione del volume del fluido circolante e ad una diminuzione della pressione sanguigna. La condizione è compensata dall'introduzione soluzioni saline e stimolatori dei recettori dell'aldosterone.
- Un tumore della corteccia surrenale porta ad un eccesso di aldosterone, che manifesta i suoi effetti e aumenta la pressione sanguigna. Vengono attivati anche i processi di divisione cellulare, si verificano ipertrofia miocardica e fibrosi e si sviluppa insufficienza cardiaca.
- Patologia epatica, quando la distruzione dell'aldosterone viene interrotta e si verifica il suo accumulo. La patologia viene trattata con bloccanti dei recettori dell'aldosterone.
- Malattie renali infiammatorie.

L'importanza del RAAS per la vita e la medicina
Sistema renina-angiotensina-aldosterone e suo ruolo nel corpo:
- accetta Partecipazione attiva nel mantenimento della pressione sanguigna normale;
- garantisce l'equilibrio di acqua e sali nel corpo;
- mantiene l'equilibrio acido-base del sangue.
Il sistema potrebbe fallire. Influenzando i suoi componenti, puoi combattere l'ipertensione. Anche il meccanismo dell’ipertensione renale è strettamente correlato al RAAS.
Gruppi di farmaci altamente efficaci sintetizzati attraverso lo studio del RAAS
- "Prily." APF. L'angiotensina I non si converte in angiotensina II. Nessuna vasocostrizione - nessun aumento della pressione sanguigna. Farmaci: Amprilan, Enalapril, Captopril, ecc. Gli ACE inibitori migliorano significativamente la qualità della vita dei pazienti con diabete, garantendo la prevenzione dell'insufficienza renale. I farmaci vengono assunti in un dosaggio minimo, che non provoca una diminuzione della pressione, ma migliora solo il flusso sanguigno locale e la filtrazione glomerulare. I medicinali sono indispensabili in caso di insufficienza renale, malattia cronica cuore e servire come uno dei mezzi per trattare l'ipertensione (se non ci sono controindicazioni).
- "Sartani". Bloccanti del recettore dell'angiotensina II. Le navi non reagiscono e non si contraggono. Farmaci: Losartan, Eprosartan, ecc.

L'opposto del sistema renina-angiotensina-aldosterone è il sistema chinina. Pertanto, il blocco del RAAS porta ad un aumento dei componenti del sistema chinina (bradichinina, ecc.) nel sangue, che ha un effetto benefico sul tessuto cardiaco e sulle pareti vascolari. Il miocardio non soffre di fame, perché la bradichinina aumenta il flusso sanguigno locale e stimola la produzione di vasodilatatori naturali nelle cellule della midollare renale e nei microciti dei dotti collettori - prostaglandine E e I2. Neutralizza l'effetto pressorio dell'angiotensina II. I vasi non subiscono spasmi, il che garantisce un adeguato apporto di sangue agli organi e ai tessuti del corpo, il sangue non viene trattenuto e la formazione di placche aterosclerotiche e coaguli di sangue viene ridotta. Le chinine hanno un effetto benefico sui reni e aumentano la diuresi (la produzione giornaliera di urina).
Renina
– un enzima sintetizzato dalle cellule iuxtaglomerulari delle arteriole afferenti renali, avente un peso molecolare di circa 40 kDa. La formazione di renina avviene in modo particolarmente intenso durante l'ischemia renale. La posizione delle cellule iuxtaglomerulari le rende particolarmente sensibili ai cambiamenti della pressione sanguigna, nonché alla concentrazione degli ioni Na+ e K+ nel fluido che scorre attraverso i tubuli renali. A causa di queste proprietà, qualsiasi combinazione di fattori che provoca una diminuzione del volume dei liquidi (disidratazione, calo della pressione sanguigna, perdita di sangue, ecc.) o una diminuzione Concentrazione di NaCl, stimola il rilascio di renina.
Allo stesso tempo, agiscono la maggior parte dei regolatori della sintesi della renina barocettori renali. Il rilascio di renina è influenzato dallo stato del sistema nervoso centrale e dai cambiamenti nella posizione del corpo nello spazio. In particolare, quando si passa dalla posizione sdraiata a quella seduta o in piedi (test clinostatico), aumenta la secrezione di renina. Questa reazione riflessa è causata da un aumento del tono della parte simpatica del sistema autonomo sistema nervoso, trasmettendo impulsi ai recettori b-adrenergici delle cellule iuxtaglomerulari.
Il substrato principale interessato dalla renina è angiotensinogeno– una proteina compresa nella frazione delle a2-globuline e prodotta dal fegato. Sotto l'influenza di glucocorticoidi ed estrogeni, la sintesi dell'angiotensinogeno aumenta in modo significativo. Come risultato dell'azione della renina, l'angiotensinogeno viene convertito in decapeptide angiotensinaIO. Questo composto ha un effetto estremamente debole e non ha un effetto significativo sui livelli di pressione sanguigna.
Nel frattempo angiotensinaIO sotto l'influenza del cosiddetto Enzima di conversione dell'angiotensina (ACE) si trasforma in un potente fattore vasocostrittore - angiotensinaII. APF(dipeptide carbossipeptidasi) è una proteina integrale situata principalmente sulla membrana delle cellule endoteliali, dell'epitelio, delle cellule mononucleari, delle terminazioni nervose, delle cellule degli organi riproduttivi, ecc. La forma solubile dell'ACE è presente in quasi tutti i fluidi corporei.
È consuetudine distinguere due isoforme di ACE. Il primo di essi ricevette il nome convenzionale di “somatico”. Questa isoforma ha un MW di 170 kDa e include domini C e N omologhi. La seconda forma di ACE (“riproduttiva”) si trova nel liquido seminale, ha un MW di circa 100 kDa e corrisponde al dominio C della prima isoforma di ACE. Ciascuno dei 2 domini indicati contiene residui amminoacidici che possono partecipare alla formazione di un legame con l'atomo di zinco. Tali strutture Zn 2+ sono tipiche di molte metalloproteinasi e sono i principali siti di interazione dell'enzima sia con il substrato che con gli ACE inibitori.
Va notato che l'ACE non porta solo alla formazione angiotensina II, ma distrugge anche bradichinina – connessione di espansione vasi sanguigni. Pertanto, l'aumento della pressione sanguigna in caso di esposizione all'ACE è associato sia alla formazione di angiotensina II che alla degradazione della bradichinina (Fig. 32).
Un ruolo importante per l'azione dell'ACE è giocato dalla composizione ionica e, in particolare, dal contenuto di ioni cloro. Pertanto, a concentrazioni elevate di Cl, il dominio C dell’ACE idrolizza sia la bradichinina che l’angiotensina I più velocemente del dominio N. Nelle regioni extracellulari, dove la concentrazione di anioni cloruro è elevata, il dominio N è principalmente responsabile della conversione dell'angiotensina-I. Tuttavia, a livello intracellulare, dove la concentrazione di CI è bassa, il dominio N può partecipare all'idrolisi di altre sostanze peptidiche.
Dietro l'anno scorsoÈ stato stabilito che l'ACE svolge un ruolo importante nell'ematopoiesi, perché sotto la sua influenza si forma peptide emopoietico, inibendo la formazione di cellule ematopoietiche nel midollo osseo.
Il ruolo dell'ACE nell'organismo è stato rivelato nei topi privi del gene ACE. Tali animali presentavano bassa pressione sanguigna, varie disfunzioni vascolari, struttura e funzione renale compromesse e infertilità nei maschi.
AngiotensinaII
aumenta la pressione sanguigna causando costrizione arteriolare ed è il più potente agente vasoattivo conosciuto. Inoltre, attraverso un meccanismo di feedback, inibisce la formazione e il rilascio di renina da parte delle cellule iuxtaglomerulari del rene, che alla fine dovrebbe ripristinare i normali livelli di pressione sanguigna. Sotto influenza angiotensinaII la produzione dei principali mineralcorticoidi aumenta notevolmente - aldosterone. Sebbene questa azione sia diretta, l’angiotensina II non influenza la produzione di cortisolo. Lo scopo principale dell'aldosterone è la ritenzione di Na+ (favorendo il suo riassorbimento nei tubuli renali) e il rilascio di K+ e H+ (principalmente attraverso i reni). Queste reazioni vengono eseguite come segue.
Aldosterone
penetra dal fluido extracellulare nel citoplasma della cellula e lì si connette con un recettore specifico, dopo di che il complesso risultante (aldosterone + recettore) penetra nel nucleo. L'aldosterone stimola anche l'apertura dei canali del Na+, consentendo agli ioni Na+ di entrare nella cellula attraverso la membrana apicale dal lume del tubulo.
L'aumento della secrezione di K + sotto l'influenza dell'aldosterone è dovuto ad un aumento della permeabilità della membrana apicale rispetto a questi ioni, grazie alla quale K + entra nel lume del tubulo dalla cellula.
La ritenzione di Na+ nel corpo, come l'angiotensina II, contribuisce all'aumento della pressione sanguigna.
AngiotensinaIIè in grado di legarsi a recettori specifici delle cellule glomerulari della ghiandola surrenale. Il contenuto di questi recettori dipende in gran parte dalla concentrazione di ioni K +. Quindi, se il livello di K + aumenta, aumenta il numero di recettori per l'angiotensina II nelle cellule glomerulari. Quando la concentrazione di ioni K+ diminuisce, si osserva l'effetto opposto. Pertanto, gli ioni K+ svolgono un ruolo importante nell'azione dell'angiotensina II sulle ghiandole surrenali.
Dietro Ultimamente lo ha determinato angiotensinaIIè in grado di attivare i macrofagi, aumentando così l'aggregazione piastrinica e accelerando la coagulazione del sangue. Allo stesso tempo, viene rilasciato inibitore dell'attivatore plasminogeno-IO (IAP-1), che può essere accompagnato da depressione della fibrinolisi. UN angiotensinaIIè uno dei fattori che contribuiscono allo sviluppo dell'aterogenesi, all'inibizione dell'apoptosi e all'aumento dello stress ossidativo nei tessuti, provocando così l'aggregazione piastrinica e la formazione di trombi.
AngiotensinaIIè in grado di migliorare la funzione miocardica, partecipa alla biosintesi della norepinefrina e di altre sostanze fisiologicamente attive. Allo stesso tempo, può agire come fattore di crescita, portando all’ipertrofia vascolare e cardiaca.
In alcuni animali e nell'uomo angiotensinaII sotto l'influenza di un enzima aminopeptidasi si converte in eptapeptide angiotensinaIII. Negli esseri umani, il livello di angiotensina II è circa 4 volte superiore a quello di angiotensina III. Entrambi questi composti influenzano la pressione sanguigna e la produzione di aldosterone e vengono scomposti abbastanza rapidamente dagli enzimi angiotensinasi.
Nelle malattie renali gravi accompagnate da ischemia, a causa dell'aumentata formazione e secrezione di renina, si osserva un aumento persistente della pressione sanguigna ( ipertensione renale). L'uso degli ACE inibitori in queste condizioni porta ad una rapida normalizzazione della pressione sanguigna.
In conclusione, va sottolineato ancora una volta che il sistema angiotensina-renina-aldosterone è strettamente correlato alla funzione del sistema callicreina-chinina, poiché la formazione dell'angiotensina II e la distruzione della bradichinina avvengono sotto l'influenza dello stesso enzima - ACE.
Catad_theme Ipertensione arteriosa- articoli
Catad_tema Obesità - articoli
Obesità e ipertensione arteriosa
Pubblicato sulla rivista:PROBLEMI DI SALUTE DELLA DONNA N. 4, volume 3, 2008
EI Astashkin, MG Glezer
Accademia medica di Mosca dal nome. I.M.Sechenova
RIEPILOGO
La revisione analizza il ruolo dell'obesità nello sviluppo dell'ipertensione arteriosa e malattia cardiovascolare, meccanismi fisiopatologici di questa connessione, il ruolo dominante del sistema renina-angiotensina-aldosterone (RAAS). Vengono discussi i problemi della correzione farmacologica dell'ipertensione nei pazienti obesi utilizzando una combinazione fissa di farmaci che bloccano il RAAS e verapamil. Viene presentata un'analisi dell'efficacia e della sicurezza dell'uso della sibutramina per la perdita di peso in pazienti con pressione alta.
Parole chiave: obesità, ipertensione arteriosa, trattamento.
ASTRATTO
Gli autori hanno analizzato il ruolo dell'obesità nello sviluppo dell'ipertensione arteriosa e delle malattie cardiovascolari, i meccanismi fisiopatologici di questa relazione e il ruolo dominante del sistema renina-angiotensina-aldosterone (RAAS). È stato dimostrato che la correzione farmacologica della pressione alta nei pazienti obesi con una combinazione fissa di bloccanti RAAS e verapamil è efficace. Viene presentata l'analisi dell'efficacia e della sicurezza della sibutramina per la perdita di peso nei pazienti con pressione alta.
Parole chiave: obesità, ipertensione arteriosa, trattamento.
L'attualità del tema in esame è dovuta al fatto che in tutto il mondo negli ultimi anni si è registrato un notevole aumento del numero di persone obese. L’obesità è attualmente considerata come uno dei principali fattori che contribuiscono allo sviluppo di malattie che rappresentano le principali cause di mortalità tra gli adulti. Prima di tutto stiamo parlando sullo sviluppo del diabete mellito di tipo 2, nonché cardiovascolare e malattie oncologiche. Un aumento di peso di 1 kg aumenta il rischio di malattie cardiovascolari del 3,1% e di diabete del 4,5-9%.
È noto che con l'obesità il rischio di sviluppare ipertensione arteriosa, fattore che influenza in modo significativo anche l'insorgenza di malattie cardiovascolari come infarti e ictus, è triplicato rispetto alle persone con peso corporeo normale. Come dimostrato nello studio INTERSALT, per ogni 4,5 kg di aumento di peso, la pressione arteriosa sistolica (PA) aumenta di 4,5 mmHg. Arte. .
L'obesità come fattore di rischio nelle donne con ipertensione arteriosa, soprattutto in età avanzata, si verifica più spesso che negli uomini. Uno dei motivi è l’ipoestrogenismo, che si verifica durante il periodo postmenopausale. Si notano alcune caratteristiche della prevalenza dell'obesità in diversi tipi di ipertensione arteriosa. Pertanto, tra le donne anziane con ipertensione sistolica isolata, l’obesità non è così comune e non esistono dati sugli effetti della perdita di peso su questa categoria di pazienti. Nelle donne con obesità addominale e forme di ipertensione arteriosa sistole-diastolica, la perdita di peso è punto importante nel controllo delle malattie.
Con l'obesità si verificano numerosi cambiamenti emodinamici, in particolare un aumento del volume sanguigno circolante, della gittata sistolica e della gittata cardiaca con resistenza vascolare relativamente normale. Si ritiene che l'ipertensione nei pazienti obesi sia dovuta principalmente all'aumento della gittata cardiaca con resistenze periferiche "non sufficientemente normali".
Questo stato emodinamico ha un effetto stimolante su due sistemi regolatori antagonisti che controllano il volume del sangue e resistenza periferica- il sistema renina-angiotensina-aldosterone (RAAS) e il sistema del peptide natriuretico cardiaco. La loro disregolazione può in gran parte spiegare l’elevata gittata cardiaca nei pazienti ipertesi obesi. Inoltre, questi sistemi di regolazione cardiovascolare sono coinvolti nei cambiamenti metabolici associati all’eccesso di peso corporeo nelle malattie cardiovascolari.
Quindi, nell’obesità, tre meccanismi principali svolgono un ruolo significativo nella patogenesi dell’ipertensione arteriosa:
La patogenesi dello sviluppo dell'ipertensione arteriosa e delle malattie cardiovascolari nell'obesità è mostrata schematicamente nella Figura 1.
Figura 1. Schema della patogenesi dell'ipertensione arteriosa e delle malattie cardiovascolari nell'obesità
Sistema renina-angiotensina-aldosterone sistemico e tissutale e suoi cambiamenti nell'obesità
Il RAAS comprende l'angiotensinogeno, la renina, l'angiotensina I, l'enzima di conversione dell'angiotensina (ACE) e l'angiotensina II (AT II). AT II ha un effetto diverso su cellule diverse che hanno recettori specifici.
Secondo i concetti classici, l'angiotensinogeno si forma nel fegato e, sotto l'influenza della renina, sintetizzata nelle cellule periglomerulari dei reni (cellule iuxtaglomerulari), l'angiotensinogeno viene convertito nel sangue in angiotensina I. L'ACE è responsabile della scissione dell'AT I, che dà luogo alla formazione di AT II.
È importante notare che nell'obesità si verifica un'interruzione dei meccanismi che regolano il funzionamento del RAAS. In condizioni fisiologiche, una maggiore attività RAAS porta ad un aumento della resistenza vasi periferici e, di conseguenza, ad un aumento della pressione sanguigna. Secondo il principio del feedback, un aumento della pressione sanguigna dovrebbe causare una diminuzione della secrezione di renina, un calo dei livelli di AT II e una diminuzione dei livelli di aldosterone. Questo, a sua volta, riduce la ritenzione di liquidi e sodio e mantiene la pressione sanguigna a livelli normali.
Tuttavia, nei pazienti con obesità viscerale, la regolazione del livello dei componenti circolanti sistemici del RAAS è compromessa. Nonostante l’aumento della pressione sanguigna, la ritenzione di sodio e di liquidi, nonché l’aumento del volume sanguigno circolante, l’attività della renina plasmatica e dell’aldosterone rimane normale o addirittura leggermente elevata. Tale disregolazione del RAAS nell’obesità può essere una conseguenza di un aumento nella formazione dei componenti RAAS e/o di un aumento secondario della loro concentrazione dovuto a difetti nel sistema del peptide natriuretico.
Si è scoperto che oltre al RAAS ematico esiste un tessuto, il cosiddetto RAAS locale, che è stato identificato in numerosi tessuti e organi, tra cui cervello, cuore, vasi sanguigni, reni, testicoli, tessuto adiposo , eccetera.
Come è noto, due fattori giocano un ruolo chiave nella formazione dell'AT II: l'attività della renina e la concentrazione dell'angiotensinogeno. Sintesi e secrezione dell'angiotensinogeno nelle cellule tipi diversi non solo determina un aumento della concentrazione locale di AT II, ma aumenta anche l’attività sistemica del RAAS. L'infusione cronica di AT II nei topi è stata accompagnata da un aumento significativo del contenuto di mRNA dell'angiotensinogeno negli adipociti. Questi risultati indicano la presenza di un feedback positivo tra AT II e angiotensinogeno, dove l'aumento del livello di un agente stimola la formazione del secondo. Nell'obesità, soprattutto di tipo viscerale, l'attività della renina plasmatica rimane, come già indicato, normale o lieve livello elevato e i livelli di angiotensinogeno e AT II aumentano.
Struttura e proprietà fisiologiche del tessuto adiposo
Il tessuto adiposo contiene diversi tipi di cellule, tra cui adipociti, macrofagi, fibroblasti, cellule endoteliali vascolari e preadipociti (adipoblasti). Quest'ultimo tipo di cellule proviene da cellule staminali pluripotenti del mesoderma. Nuovi adipociti differenziati (“piccoli”) si formano dai preadipociti nel corpo umano adulto. Questi adipociti aumentano di dimensioni (adipociti “grandi”) a causa del maggiore apporto alimentare acidi grassi. Gli acidi grassi a catena lunga entrano negli adipociti dal sangue e si depositano sotto forma di triacilgliceroli neutri. Il tessuto adiposo è responsabile dell'immagazzinamento e della secrezione di acidi grassi a catena lunga, che fungono da uno dei principali substrati energetici per molti organi e tessuti, ad esempio i muscoli cardiaci e scheletrici. Gli adipociti “più grandi” secernono una quantità significativamente maggiore di acidi grassi saturi. L'idrolisi dei trigliceridi e il rilascio degli acidi grassi avvengono sotto l'influenza della lipasi intracellulare sensibile agli ormoni, la cui attività è controllata dalle catecolamine (regolazione positiva) e dall'insulina (regolazione negativa).
Attività endocrina del tessuto adiposo
A differenza di Grasso sottocutaneo, che tipicamente rappresenta il 75% del tessuto adiposo totale del corpo ed è il principale sito di stoccaggio dei lipidi, il grasso viscerale è attualmente considerato un tessuto attivo produttore di ormoni.
Si producono gli adipociti vasta gamma ormoni e citochine coinvolti nel metabolismo del glucosio (adiponectina, resistina, ecc.), dei lipidi (proteina di trasferimento degli esteri del colesterolo), dell'infiammazione (TNF-α, interleuchina-6), della coagulazione (inibitore dell'attivatore del plasminogeno-1), della regolazione della pressione sanguigna (angiotensinogeno, AT II), comportamento alimentare(leptina), oltre ad influenzare il metabolismo e l'attività funzionale di vari organi e tessuti, inclusi muscoli, fegato, cervello e vasi sanguigni (vedi tabella).
Tavolo. Funzione endocrina adipociti: adipocitochine
| Adipocitochine | Effetti delle adipocitochine |
| Leptina | Assorbimento degli alimenti, massa grassa |
| Adiponectina | |
| Resistere | Resistenza all'insulina, infiammazione |
| Visfatina | Resistenza all'insulina |
| Omentino | Resistenza all'insulina |
| Serpina rilasciata dal tessuto adiposo viscerale (Vaspin) | Resistenza all'insulina |
| Apelin | Vasodilatazione |
| Proteina di trasferimento degli esteri del colesterolo (CETP) | Metabolismo dei lipidi |
| Lipasi lipoproteica (LPL) | Metabolismo dei lipidi |
| Lipasi sensibile agli ormoni (HSL) | Metabolismo dei lipidi |
| Proteina-4 legante gli acidi grassi degli adipociti (A-FABP-4 (aP2)) | Metabolismo dei lipidi |
| Perlipino | Metabolismo dei lipidi |
| Proteina legante il renitolo (RBP) | Metabolismo dei lipidi |
| Proteina stimolante l'acilazione (ASP) | Metabolismo dei lipidi |
| Angiotensina II (AT II) | Pressione arteriosa |
| Enzima di conversione dell'angiotensina (ACE) | Pressione arteriosa |
| Angiotensinogeno (AGT) | Pressione arteriosa |
| Fattore di necrosi tumorale alfa (TNF-a) | Infiammazione |
| Interleuchina, 6 (IL-6) | Infiammazione |
| Proteina C-reattiva (PCR) | Infiammazione |
| Adipociti-tripsina/fattore del complemento D (Adipsina) | Infiammazione |
| Proteina chemioattrattiva dei macrofagi-1 (MCP-1) | Attrattivo per i macrofagi |
| Molecola di adesione intercellulare-1 (ICAM-1) | Attivazione dei macrofagi |
| Inibitore dell'attivatore del plasminogeno-1 (PAI-1) | Fibrinolisi |
È importante sottolineare che anche un piccolo aumento di volume Grasso viscerale svolge un ruolo significativo nei disordini metabolici, nella regolazione dell'equilibrio idrico-elettrolitico e nelle malattie cardiovascolari.
Con l'aumento della massa del tessuto adiposo aumenta il contenuto di quasi tutte le adipochine nel sangue. L'eccezione è l'adiponectina, il cui livello rientra in queste condizioni. Leptina e adiponectina sono le adipochine attualmente più studiate.
Leptina. La produzione di leptina avviene principalmente negli adipociti “grandi”. La leptina è spesso considerata una molecola di segnalazione che media la relazione tra il contenuto di nutrienti che entrano nel corpo, lo stato del tessuto adiposo e il sistema nervoso centrale (ipotalamo). La leptina aumenta l'ossidazione dei lipidi nel fegato, così come la lipolisi negli adipociti e muscoli scheletrici. L'insulina stimola la formazione di leptina. I livelli di leptina sono influenzati anche dagli acidi grassi liberi, dal TNF-α, dagli estrogeni e dall’ormone della crescita.
Adiponectina. La produzione di adiponectina avviene esclusivamente negli adipociti. L'adiponectina ha una varietà di effetti biologici: ha un effetto antiaterogenico, aumenta la sensibilità cellulare all'insulina, sopprime la sintesi del glucosio nel fegato, migliora il suo trasporto ai muscoli e aumenta l'ossidazione degli acidi grassi. I livelli di adiponectina sono ridotti nell’obesità, nella resistenza all’insulina e nel diabete di tipo 2.
Tessuto adiposo e attività RAAS
Si è scoperto che il tessuto adiposo è al secondo posto dopo il fegato nella formazione di angiotensinogeno. Ad esempio, la quantità di mRNA dell’angiotensinogeno negli adipociti è circa il 70% del livello nel fegato. La relazione tra livelli di angiotensinogeno, obesità e ipertensione arteriosa è stata chiaramente dimostrata in esperimenti su topi transgenici che esprimono quantità eccessive di angiotensinogeno nel tessuto adiposo. Questi topi presentano obesità viscerale e ipertensione. Preadipociti e differenziati cellule adipose possiedono un set completo di componenti necessari per la sintesi locale di AT II, nonché il recettore AT 1 per AT II, che garantisce la trasmissione intracellulare dei segnali di attivazione innescati da AT II. Nell’obesità il volume degli adipociti differenziati viscerali aumenta di 20-30 volte. L'obesità è caratterizzata da una disfunzione degli adipociti, intesa come aumento della formazione e secrezione di varie adipochine, citochine, nonché un aumento del contenuto dei componenti RAAS, principalmente nel grasso viscerale.
Riassumendo i dati di vari studi, possiamo affermare che con l'obesità si verifica un aumento dell'attività del RAAS, che si riflette nei seguenti fatti:
Alta attività Il RAAS, a sua volta, porta ad un aumento della massa del tessuto adiposo. Nello specifico, i topi transgenici che sovraesprimevano l’angiotensinogeno solo nelle cellule adipose hanno mostrato un aumento dei livelli di angiotensinogeno nel sangue, sviluppo di ipertensione e aumento della massa del tessuto adiposo. Il tessuto AT II funziona essenzialmente come fattore di crescita per gli adipociti. AT II, a causa del suo effetto sui recettori AT 1, provoca un aumento della proteina ciclina D 1, che è coinvolta nella regolazione della crescita e della divisione delle cellule adipose. È stato dimostrato che AT II induce il passaggio della fase G 1 ciclo cellulare nei preadipociti umani. Questo effetto è stato associato ad un effetto sui recettori AT 1 e alla successiva attivazione della chinasi ciclina D 1-dipendente.
È stato stabilito che AT II provoca la differenziazione dei preadipociti, attiva gli enzimi chiave della formazione dei lipidi (lipogenesi) e aumenta l'accumulo di trigliceridi negli adipociti.
L'obesità viscerale è accompagnata da un aumento dell'attività della 11-beta-idrossisteroide deidrogenasi di tipo 1, che porta alla formazione di cortisolo, ormone chiave nella differenziazione dei preadipociti in adipociti.
L'attività del RAS tissutale è strettamente correlata alla produzione di adipochine da parte del tessuto adiposo. Ad esempio, è stato dimostrato che AT II induce l’espressione della leptina negli adipociti. È stato suggerito che tale attività sia caratteristica solo dell'AT II sintetizzato localmente, in contrasto con l'AT II sistemico.
Obesità e attività del sistema nervoso simpatico
Nell'obesità, soprattutto nella sua variante addominale, si osserva molto spesso l'attivazione del sistema nervoso simpatico. Il NAS (Normotesive Aging Study) ha riscontrato un aumento della norepinefrina urinaria proporzionale all’indice di massa corporea. Man mano che si perde peso, l’attività del sistema nervoso simpatico diminuisce.
L’aumento dell’attività del sistema nervoso simpatico nell’obesità è facilitato dalla presenza di iperinsulinemia e resistenza all’insulina. L’insulina può aumentare da sola l’attività del sistema simpatico-surrenale, ma ciò potrebbe essere dovuto in parte all’azione della leptina. È noto che all’aumentare del grado di obesità aumenta il livello di leptina a digiuno, secreta dagli adipociti. La leptina aumenta l'attività del sistema nervoso simpatico, soprattutto nei reni. Ciò porta, da un lato, ad un rendimento elevato e ad un aumento della frequenza cardiaca e, dall'altro, ad un aumento del riassorbimento del sodio e ad un aumento del volume del sangue intravascolare.
È stata stabilita una relazione tra il RAAS e il sistema nervoso simpatico. L'attivazione del sistema nervoso simpatico è associata ad un aumento della secrezione di renina nei reni e ciò avviene indipendentemente dalla concentrazione intrarenale. sistema sensoriale regolare la secrezione di renina da parte dei reni. Inoltre, un aumento dell'adenosina monofosfato ciclico sotto l'influenza delle catecolamine stimola l'espressione dell'angiotensinogeno negli adipociti umani. Un aumento dei livelli di AT II aumenta l'attività del sistema nervoso simpatico nell'uomo. È stato stabilito che AT II attiva il sistema nervoso simpatico locale, che è coinvolto nell'aumento della temperatura corporea (termogenesi). Il trattamento a freddo porta ad un aumento del contenuto di AT II negli adipociti senza un concomitante cambiamento dei livelli plasmatici di AT II.
Pertanto, la disregolazione del RAAS nell’obesità può anche stimolare l’attività del sistema nervoso simpatico.
Metodi di correzione farmacologica dell'ipertensione arteriosa nell'obesità
Il contributo dei diversi meccanismi patogenetici al mantenimento dell’ipertensione arteriosa nell’obesità può essere diverso. Pertanto, farmaci antipertensivi con meccanismi d’azione molto diversi possono avere un effetto benefico in questa situazione.
In conformità con le moderne raccomandazioni per il trattamento dell'ipertensione arteriosa, la chiave per il successo di una significativa riduzione della pressione sanguigna è l'uso della terapia combinata. Per i pazienti obesi, innanzitutto, i componenti principali di tale terapia dovrebbero contenere una combinazione di farmaci che riducono l'attività dei RAAS (ACE inibitori e sartani), con farmaci che riducono l'attività del sistema nervoso simpatico (β-bloccanti e calcioantagonisti non diidropiridinici) e diuretici. Alta efficienza L’uso di farmaci che bloccano il RAAS nell’obesità è stato dimostrato in molti studi. Per quanto riguarda l’uso dei β-bloccanti, i dati sono molto contraddittori, innanzitutto a causa dei dubbi sulla loro utilità per il trattamento di pazienti con ipertensione arteriosa non complicata, e in secondo luogo perché i β-bloccanti, almeno quelli classici, possono aumentare il peso dei pazienti e aumentare la resistenza all'insulina. Pertanto, se scegliamo i β-bloccanti per il trattamento di pazienti con obesità o sindrome metabolica, allora dovrebbero essere farmaci con proprietà speciali, in particolare carvedilolo e nebivololo.
Allo stesso tempo, si è scoperto che il calcioantagonista non diidropiridinico verapamil può non solo ridurre significativamente la pressione sanguigna, ma anche ridurre l'attività del sistema nervoso simpatico.
Pertanto, in caso di obesità, una combinazione di farmaci che bloccano il RAAS e verapamil può essere utilizzata per trattare l’ipertensione arteriosa.
Va sottolineato che questo tipo di combinazione medicinali esiste sotto forma di una combinazione già pronta forma di dosaggio- il farmaco Tarka, che contiene un ACEI liposolubile - trandolapril e verapamil a lento rilascio (verapamil SR). Questo approccio è molto importante per una terapia efficace, poiché l'uso di forme di dosaggio finite migliora l'aderenza del paziente al trattamento.
Esistono prove che Tarka, in misura maggiore di ciascuno dei suoi componenti, riduce la pressione sanguigna, ha una spiccata capacità di ridurre l'ipertrofia ventricolare sinistra, aiuta a normalizzare la funzione endoteliale ed è metabolicamente neutro, anche nei pazienti con diabete mellito.
Doppia azione- riduzione dell'attività del RAAS sotto l'influenza di trandolapril e del sistema nervoso simpatico a causa dell'azione prolungata del verapamil - forniscono un effetto importante su meccanismi patogenetici lo sviluppo dell'ipertensione arteriosa nell'obesità e i meccanismi che provocano il danno agli organi bersaglio in questo tipo di ipertensione.
Particolare attenzione, quando si discute del trattamento dell'ipertensione nell'obesità, dovrebbe essere prestata al fatto che la terapia basata su una combinazione di trandalapril con verapamil a lunga durata d'azione può ridurre il rischio di sviluppare diabete mellito rispetto all'uso di un'altra tattica terapeutica - una combinazione di sartan con una bassa dose di un diuretico tiazidico. Lo studio STAR mostra chiaramente che quando Tarka viene utilizzato per un anno, meno persone con sindrome metabolica, in cui l’obesità addominale è una condizione predominante, sviluppano diabete(Fig. 2) .
Figura 2. Sviluppo di nuovi casi di diabete mellito (glucosio a digiuno > 126 mg/dl o test di tolleranza al glucosio a 2 ore > 200 mg/dl) per tipo di terapia antipertensiva nei soggetti con sindrome metabolica nello studio STAR

Inoltre, secondo lo studio STAR-LET, anche quando il diabete mellito si manifesta in sottofondo terapia farmacologica il trasferimento di questi pazienti al farmaco Tarka ha permesso la normalizzazione nella metà dei pazienti metabolismo dei carboidrati.
I risultati di questi studi ci costringono a riconsiderare le raccomandazioni per terapia farmacologica ipertensione arteriosa nei soggetti con sindrome metabolica e iniziare la terapia con una combinazione contenente un ACE inibitore (o sartan) e un calcio antagonista, oppure trasferire i pazienti a una terapia simile.
Come è stato più volte menzionato, per il trattamento dell'ipertensione arteriosa gioca un ruolo importante la riduzione del peso dei pazienti e del grado di obesità addominale. Naturalmente, ridurre il peso corporeo in un modo o nell’altro può avere un impatto significativo sulla riduzione dell’incidenza delle malattie cardiovascolari. Attualmente esistono diversi approcci per la terapia farmacologica dell’obesità. Il primo è trattamento sintomatico, vale a dire, ridurre il numero di calorie consumate riducendo l'assorbimento dei grassi dal cibo. Questo approccio può essere chiamato compensativo. Infatti, con tale terapia la malattia non viene eliminata (poiché il paziente continua a mangiare troppo), ma viene compensata solo temporaneamente dal farmaco. Un altro approccio per trattare il sovrappeso e l’obesità consiste nell’affrontare la radice del problema, vale a dire l’eccesso di cibo cronico. Ecco come funziona la sibutramina (Meridia). Porta ad una rapida sazietà e riduce la quantità di cibo consumato sopprimendo la ricaptazione della norepinefrina e della serotonina nelle sinapsi dei circuiti neuronali. Oggi Meridia è l'unica farmaco originale, eliminando la causa dell'obesità.
La differenza fondamentale tra la sibutramina è che, senza provocare una diminuzione dell'appetito, favorisce una più precoce sensazione di sazietà. Una persona si libera dell'abitudine patologica di mangiare troppo, che si traduce in una diminuzione graduale e duratura del peso corporeo. Sotto l'influenza della sibutramina, il consumo di cibo si riduce di circa il 20%. Insieme a questo, la sibutramina influenza indirettamente il livello delle ammine biogene nel sangue, che attivano i recettori adrenergici nel tessuto adiposo e avviano la lipolisi negli adipociti, che è accompagnata da un cambiamento nel contenuto dei substrati energetici nel sangue. La sibutramina, grazie all'attivazione dei recettori adrenergici β 2 e β 3, migliora i processi di termogenesi e aumenta il consumo di energia nel corpo.
L'efficacia clinica e la sicurezza della sibutramina (Meridia) sono state dimostrate in grandi quantità studi multicentrici. In particolare, nello studio STORM (Sibutramine Trial on Obesity Reduction and Maintenance), che ha coinvolto 605 pazienti obesi, è stato dimostrato che l'uso di sibutramina per due anni ha ridotto il peso dei pazienti di 3 volte e la circonferenza della vita - 2 volte più pronunciata rispetto al placebo... È importante sottolineare che l’80% dei pazienti ha mantenuto la perdita di peso per due anni, rispetto al 16% dei pazienti che hanno ricevuto placebo (p< 0,001). Показательно, что при этом улучшался липидный спектр: уровень липопротеидов alta densità aumentato del 21% con una diminuzione dei livelli di lipoproteine a bassa densità e trigliceridi.
L’effetto benefico della perdita di peso nel trattamento di pazienti con ipertensione arteriosa e altre malattie cardiovascolari potrebbe risiedere anche nel fatto che una diminuzione del grasso intra-addominale può ridurre la compressione meccanica dei reni, il che può portare ad un miglioramento dell’afflusso di sangue ai reni. e una diminuzione dell'attività del RAAS. La riduzione del tessuto adiposo all'interno e attorno ai reni può portare a una diminuzione della pressione interstiziale, alla compressione della porzione sottile dell'ansa di Henley, a un aumento del flusso sanguigno nei vasa recta e a una diminuzione del riassorbimento tubulare di Na+ e acqua. Pertanto, la perdita di peso dovuta a metodi di correzione non farmacologici o farmacologici può ridurre la pressione sanguigna.
Tuttavia, fino a poco tempo fa, in realtà pratica clinica la sibutramina è stata usata con cautela, temendo che fosse possibile influenza negativa sulla pressione arteriosa e sulla frequenza cardiaca, il che, a sua volta, potrebbe comportare, seppure in un numero limitato di pazienti, risultati spiacevoli sentimenti soggettivi. Per studiare l’effetto della sibutramina sul sistema cardiovascolare e dimostrare la sicurezza del farmaco in un gruppo di pazienti con un aumentato rischio di malattie cardiovascolari, è stato condotto uno studio internazionale multicentrico su larga scala, in doppio cieco, controllato con placebo, SCOUT (Sibutramine Cardiovascolare). OUTcomes), in cui sono stati osservati 10.742 pazienti, di cui il 97% presentava patologie del sistema cardiovascolare, 88 - ipertensione arteriosa e 84% - diabete mellito di tipo 2. Sulla base dei risultati della prima fase completata dello studio, è stato riscontrato che la somministrazione di sibutramina ha portato ad un significativo (p< 0,001) уменьшению веса (медиана изменения составила 2,2 кг), окружности талии (на 2 см в равной степени выраженному у мужчин и женщин) и снижению АД систолического на 3,0 мм рт. ст. и диастолического - на 1,0 мм рт. ст. Частота сердечных сокращений увеличивалась в среднем на 1,5 удара в минуту. Увеличение АД и увеличение частоты пульса наблюдалось соответственно у 4,7 и 3,5% пациентов. Таким образом, в данном исследовании было показано, что даже у пациентов, относящихся к группам высокого риска, применение сибутрамина (препарата Меридиа) было высокоэффективным и безопасным . Дальнейший анализ данных исследования SCOUT позволил установить, что у пациентов с артериальной гипертонией снижение АД при приеме сибутрамина было более выраженным и составило в среднем для систолического АД -6,5 (-27,0; 8,0) мм рт. ст., а для диастолического -2,0 (-15,0; 8,0) мм рт. ст. (p < 0,001). Среди пациентов, у которых снижение веса не было выраженным, снижение АД было достоверным, но менее выраженным, чем у лиц с успешным снижением веса, и составило в среднем для систолического -3,5 (-26,0; 10,0) мм рт. ст. и -1,5 (-16,0; 9,0) мм рт. ст. для диастолического АД (p < 0,001). У лиц с нормальным АД было достоверное, но не выраженное увеличение АД - 1,5 (-15,0; 19,5) мм рт. ст. систолического и на 1,0 (-10,5; 13,0) мм рт. ст. диастолического АД (p < 0,001). Степень повышения АД при приеме сибутрамина уменьшалась в соответствии со степенью потери веса .
Sorge spontanea la domanda su come questo o quel tipo di terapia antipertensiva si relazionerà al trattamento con sibutramina. Sono stati condotti diversi studi per rispondere a questa domanda. Ad esempio, è stato dimostrato che l'uso di una forma di dosaggio combinata contenente verapamil 180 mg/trandolapril 2 mg in combinazione con sibutramina 10 mg ha portato nell'arco di 6 mesi a una diminuzione più pronunciata della pressione sanguigna rispetto alla sola terapia antipertensiva: la pressione sanguigna sistolica è diminuita, di conseguenza, di 21,9 ± 8,1 contro 15,9 ± 12,3 mm Hg. Arte. e diastolico - di 15,7 ± 8,1 contro 9,1 ± 9,9 mm Hg. Arte. (p = 0,03). La terapia di combinazione ha portato anche ad un miglioramento più pronunciato dei parametri antropometrici; affidabile (pag<5) по сравнению с исходным уровнем снижение малых липопротеидов низкой плотности, С-реактивного белка и висфатина наблюдалось только в группе пациентов, получавших комбинированную терапию сибутрамином с антигипертензиным препаратом Тарка .
Lo studio HOS (Hypertension-Obesity-Sibutramine) era uno studio prospettico, multicentrico, controllato con placebo, in doppio cieco, della durata di 16 settimane, che confrontava diversi regimi antipertensivi (felodipina 5 mg/ramipril 5 mg (n = 57), verapamil 180 mg/ trandolapril 2 mg (n = 55), metoprololo succinato 95 mg/idroclorotiazide 12,5 mg n = 59) quando prescritti sibutramina e placebo. Questo studio ha confermato che la sibutramina può aumentare la pressione sanguigna. Pertanto, ovviamente, è necessaria un'adeguata terapia antipertensiva durante il periodo di utilizzo della sibutramina nei pazienti con ipertensione arteriosa. È stato inoltre dimostrato che, quando trattata con una combinazione di un β-bloccante e idroclorotiazide, gli effetti positivi della sibutramina in termini di perdita di peso, circonferenza della vita e l'effetto sul profilo metabolico erano significativamente meno pronunciati rispetto a quando combinata con la terapia di combinazione con ACE. inibitori e calcioantagonisti con sibutramina. Ciò conferma ancora una volta la necessità di un'attenta selezione della terapia antipertensiva nei pazienti obesi, soprattutto quando si conducono programmi mirati alla perdita di peso. E in conclusione, va notato che dal nostro punto di vista, uno dei problemi significativi che riduce l'efficacia della lotta contro l'obesità è che né i medici né la popolazione considerano l'obesità un fattore di rischio significativo. Inoltre, i pazienti spesso non si valutano obesi. Nello studio POLONESE, ad esempio, secondo la valutazione dei medici basata sul calcolo del BMI, l'obesità sia negli uomini che nelle donne è stata rilevata tre volte più spesso rispetto all'autovalutazione dei pazienti. Pertanto, il lavoro di sensibilizzazione dovrebbe essere rafforzato e portato avanti tra la popolazione sulla necessità di prevenire l'aumento di peso, correggere l'obesità esistente e l'importanza del trattamento costante dell'ipertensione arteriosa. LETTERATURA
1. Flegal K.M., Carroll M.D., Ogden C.K., Johnson C.L. Prevalenza e tendenze dell'obesità tra gli adulti statunitensi, 1999-2000. JAMA2002; 288:1723-7.
2. Pi-Sunyer F.X. L'epidemiologia della distribuzione centrale del grasso in relazione alla malattia. Nutr Rev 2004; 62(7): 120-6.
3. Arbeeny C.M. Affrontare l’esigenza medica insoddisfatta di terapie dimagranti sicure ed efficaci. Obes Res 2004; 12(8): 1191-6.
4. Stamler J., Rose G., Stamler R., et al. Risultati dello studio INTERSALT. Implicazioni sulla sanità pubblica e sull’assistenza sanitaria. Ipertensione 1989; 14(5): 570-7.
5. Jedrychowski W., Mroz E., Bojanczyk M., Jedrychowska I. Peso eccessivo e ipertensione negli anziani: i risultati dello studio comunitario. Arco Gerontol Geriatr 1991; 13(1): 61-9.
6. Kanai H., Tokunaga K., Fujioka S., et al. La diminuzione del grasso viscerale intra-addominale può ridurre la pressione sanguigna nelle donne ipertese obese. Ipertensione 1996; 27(1): 125-9.
7. Poirier P., Giles T.D., Bray G.A., et al. Associazione americana del cuore; Comitato sull'obesità del Consiglio su nutrizione, attività fisica e metabolismo. Obesità e malattie cardiovascolari: fisiopatologia, valutazione ed effetto della perdita di peso: un aggiornamento della Dichiarazione scientifica dell'American Heart Association del 1997 sull'obesità e le malattie cardiache del Comitato sull'obesità del Consiglio su nutrizione, attività fisica e metabolismo. Circolazione 2006; 113:898-918.
8.Alpert M.A. Cardiomiopatia da obesità; Fisiopatologia ed evoluzione della sindrome clinica. Am J Med Sci 2001; 321: 225-36.
9. Aneja A., El-Atat F., McFarlane S.I., Sowers J.R. Ipertensione e obesità. Recente Progr Horm Res 2004; 59: 169-205.
10. Engeli S., Sharma A.M. Il sistema renina-angiotensina e i peptidi natriuretici nell’ipertensione associata all’obesità. J Mol Med 2001; 79: 21-9.
11. Lafontan M., Moro C., Sengenes C., et al. Un ruolo metabolico insospettato per i peptidi natriuretici atriali: il controllo della lipolisi, della mobilizzazione dei lipidi e dei livelli sistemici di acidi grassi non esterificati nell'uomo. Trombo Arteriosclero Vasc Biol 2005; 25: 2032-42.
12. Sala J.E. Il rene, l'ipertensione e l'obesità. Ipertensione 2003; 41(3): 625-33.
13. Cooper R., McFarlane Anderson N., Bennet F. I., et al. ACE, angiotensinogeno e obesità: una potenziale via che porta all’ipertensione. J Hum Hypertens 1997; 11: 107-11.
14. Lu N., Boustany-Kari C.M., Daugherty A., Cassis L.A. L’angiotensina II aumenta l’espressione dell’angiotensinogeno adiposo. Am J Physiol Endocrinol Metab 2007; 292: 1280-7.
15. Engeli S., Negrel R., Sharma A.M. Fisiologia e fisiopatologia del sistema renina-angiotensina del tessuto adiposo. Ipertensione 2000; 35(6): 1270-7.
16. Otto T.C., Lane M.D. Lo sviluppo adiposo: dalla cellula staminale all'adipocita. Crit Rev Biochem Mol Biol 2005; 40: 229-42.
17. Hajer G.R., van Haeften T.W., Visseren F.L.J. Disfunzione del tessuto adiposo nell’obesità, nel diabete e nelle malattie vascolari. Eur Cuore J 2008; 29: 2959-71.
18. Wannathee S. G., Lowe G. D., Rumley A., et al. Adipochine e rischio di diabete di tipo 2 negli uomini anziani. Diabete Core 2007; 30: 1200-5.
19. Chu N.F., Spiegelman D., Hotamisligil G.S., et al. Livelli plasmatici di insulina, leptina e recettori solubili del TNF in relazione ai fattori di rischio di malattie cardiovascolari aterogene e trombogeniche correlate all’obesità negli uomini. Ateroslerosi 2001; 157: 495-503.
20. Skurk T., Alberti-Huber C., Herder C., Hauner H. Relazione tra dimensione degli adipociti ed espressione e secrezione di adipochine. J Clin Endocrinol Metab 2007; 6(92): 1023-33.
21. Ran J, Hirano T, Fukui T, et al. L'infusione di angiotensina II diminuisce il livello plasmatico di adiponectina attraverso il suo recettore di tipo 1 nei ratti: un'implicazione per la resistenza all'insulina correlata all'ipertensione. Metabolismo 2006; 55: 478-88.
22. Considine R.V., Sinha M.K., Heiman M.L., et al. Concentrazione sierica di leptina immunoreattiva in esseri umani normopeso e obesi. N Engl J Med 1996; 334:292-5.
23. Schwartz M.W., Woods S.C., Porte D. Jr., et al. Controllo del sistema nervoso centrale sull'assunzione di cibo. Natura 2000; 404(6778): 661-71.
24. Cheung C.C., Clifton D.K., Steiner R.A. I neuroni proopiomelanocortina sono bersagli diretti per la leptrina nell'ipotalamo. Endocrinologia 1997; 138; 4489-92.
25. Lungo Y.C., Zierath J.R. Segnalazione della proteina chinasi attivata da AMP nella regolazione metabolica. J Clin Investire 2006; 116: 1776-83.
26. Saladin R., De Vos P., Guerre-Millo M., et al. Aumento transitorio dell’espressione genica dell’obesità dopo l’assunzione di cibo o la somministrazione di insulina. Natura 1995; 377:527-9.
27. Zhang H.H., Kumar S., Barnet A.H., Eggo M.C. Il fattore di necrosi tumorale alfa esercita un duplice effetto sulla sintesi e sul rilascio della leptina adiposa umana. Mol Cell Endocrinol 2000; 159: 79-88.
28. Lindsay R.S., Funahashi T., Hanson R.L., et al. Adiponectina e sviluppo del diabete di tipo 2 nella popolazione indiana Pima. Lancetta 2002; 360: 57-8.
29. Hajer G.R., van der Graaf Y., Olijhoek J.K., et al. Bassi livelli plasmatici di adiponectina sono associati a un basso rischio di futuri eventi cardiovascolari in pazienti con malattia vascolare clinicamente evidente. Sono il cuore J 2007; 154(750): 1-7.
30. Massiera F., Bloch-Faure M., Ceiler D., et al. L’angiotensinogeno adiposo è coinvolto nella crescita del tessuto adiposo e nella regolazione della pressione sanguigna. FASEB J 2001; 15: 2727-9.
31. Paul M., Mehr A.P., Kreutz R. Fisiologia dei sistemi renina-angiotensina locali. Physiol Rev 2006; 86: 747-803.
32. Karlsson C., Lindell K., Ottosson M., et al. Il tessuto adiposo umano esprime l'angiotensinogeno e gli enzimi necessari per la sua conversione in angiotensina II. J Clin Endocrinol Metab 1998; 83: 3925-9.
33. Gorzelniak K., Engeli S., Janke J., et al. Regolazione ormonale del sistema renina-angiotensina del tessuto adiposo umano: relazione con l'obesità e l'ipertensione. J Hypertens 2002; 20: 965-73.
34. Engeli S., Gorzelniak K., Kreutz R., et al. Co-espressione dei geni del sistema renina-angiotensina nel tessuto adiposo umano. J Hypertens 1999; 17: 555-60.
35. Achard V., Boullu-Ciocca S., Desbrière R., et al. Espressione del recettore della renina nel tessuto adiposo umano. Am J Physiol Regul Integr Comp Physiol 2007; 292: 274-82.
36. Sarzani R., Savi F., Dessi-Fulgheri P., Rappelli A. Sistema renina-angiotensina, peptidi natriuretici, obesità, sindrome metabolica e ipertensione: una visione integrata nell'uomo. J Hypertens 2008; 26: 831-43.
37. Crandall D. L., Armellino D. C., Busler D. E., et al. Recettori dell'angiotensina II nei preadipociti umani: ruolo nella regolazione del ciclo cellulare. Endocrinologia 1999; 140: 154-8.
38. Saint-Marc P., Kozak L.P., Ailhaud G., et al. L'angiotensina II come fattore trofico del tessuto adiposo bianco: stimolazione della formazione delle cellule adipose. Endocrinologia 2001; 142: 487-92.
39. Watanabe G., Lee R. J., Albanese C., et al. Attivazione dell'angiotensina II dell'attività della chinasi ciclina D1-dipendente. J Biol Chem 1996; 271:22570-7.
40. Darimont C., Vassaux G., Ailhaud G., Negrel R. Differenziazione delle cellule preadipose: ruolo paracrino della prostaciclina dopo stimolazione delle cellule adipose da parte dell'angiotensina-II. Endocrinologia 1994; 135:2030-6.
41. Jones B.H., Standridge M.K., Moustaid N. L'angiotensina II aumenta la lipogenesi nelle cellule 3T3-L1 e adipose umane. Endocrinologia 1997; 138: 1512-9.
42. Wake DJ, Walker B.R. Inibizione dell'11beta-idrossisteroide deidrogenasi di tipo 1 nell'obesità. Endocrino 2006; 29: 101-8.
43. Engeli S., Bohnke J., Feldausch M., et al. Regolazione dei geni 11beta-HSD nel tessuto adiposo umano: influenza dell'obesità centrale e della perdita di peso. Obes Res 2004; 12:9-17.
44. Kim S., Whelan J., Claycombe K., et al. L’angiotensina II aumenta la secrezione di leptina da parte di 3T3-L1 e degli adipociti umani attraverso un meccanismo indipendente dalle prostaglandine. J Nutr 2002; 132: 1135-40.
45. Cassis L.A., English V.L., Bharadwaj K., Boustany C.M. Effetti differenziali dell'angiotensina II locale rispetto a quella sistemica nella regolazione del rilascio di leptina dagli adipociti. Endocrinologia 2004; 145: 169-74.
46. Mancia G., Bousquet P., Elghozi J.L., et al. Il sistema nervoso simpatico e la sindrome metabolica. J Hypertens 2007; 25: 909-20.
47. Tentolouris N., Liatis S., Katsilambros N. Attività del sistema simpatico nell'obesità e nella sindrome metabolica. Ann NY Acad Sci 2006; 1083: 129-52.
48. Landsberg L., Troisi R., Parker D., et al. Obesità, pressione sanguigna e sistema nervoso simpatico. Ann Epidemiol 1991; 1: 295-303.
49. Serazin V., Dos Santos E., Morot M., Giudicelli Y. L'espressione e la secrezione del gene dell'angiotensinogeno adiposo umano sono stimolate dall'AMP ciclico attraverso una maggiore attività di legame dell'elemento reattivo all'AMP ciclico del DNA. Endocrino 2004; 25: 97-104.
50. Cassis L.A. Ruolo dell'angiotensina II nella termogenesi dell'adiposo bruno durante l'acclimatazione al freddo. Am J Physiol Endocrinol Metab 1993; 265:860-5.
51. Cassis L.A., Dwoskin L.P. Modulazione presinaptica del rilascio di neurotrasmettitori da parte dell'angiotensina II endogena nel tessuto adiposo bruno. J Neural Transm 1991; 34: 129-37.
52. Reisin E., Weir M., Falkner B., et al. Lisinopril contro idroclorotiazide in pazienti ipertesi obesi: uno studio multicentrico controllato con placebo. Gruppo di studio Trattamento in pazienti obesi con ipertensione (TROPHY). Ipertensione 1997; 30(1): 2140-5.
53. Neutel J.M., Saunders E., Bakris G.L., et al. L'efficacia e la sicurezza delle combinazioni fisse a basso e alto dosaggio di irbesartan/idroclorotiazide in pazienti con pressione arteriosa sistolica non controllata in monoterapia: lo studio INCLUSIVE. J Clin Hypertens (Greenwich) 2005; 7(10): 578-86.
54. Belenkov Yu.N., Chazova I.E., Mychka V.B. per conto del gruppo di ricerca IVF. Uno studio multicentrico, randomizzato, in aperto che esamina l'efficacia dei cambiamenti dello stile di vita e della terapia con ACE inibitori (quinapril) in pazienti obesi con ipertensione arteriosa (IVF). Ipertensione arteriosa 2003; 9(6): 3-6.
55. Jacob S., Rett K., Henriksen E.J. Terapia antipertensiva e sensibilità insulinica: dobbiamo ridefinire il ruolo dei beta-bloccanti? Am J Hypertens 1998; 11(10): 1258-65.
56. Kaaja R, Kujala S, Manhem K, et al. Effetti della terapia simpaticolitica sugli indici di sensibilità all'insulina nelle donne ipertese in postmenopausa. Int J Clin Pharmacol Ther 2007; 45(7): 394-401.
57. Kuperstein R., Sasson Z. Effetti della terapia antipertensiva sul metabolismo del glucosio e dell'insulina e sulla massa ventricolare sinistra: uno studio randomizzato, in doppio cieco, controllato su 21 ipertesi obesi. Circolazione 2000; 102(15): 1802-6.
58. Halperin A.K., Cubeddu L.X. Il ruolo dei calcioantagonisti nel trattamento dell’ipertensione. Sono il cuore J 1986; 111(2): 363-82.
59. McAllister R.G. Jr. Farmacologia clinica degli agenti bloccanti dei canali lenti. Prog Cardiovasc Dis 1982; 25(2): 83-102.
60. Binggeli C., Corti R., Sudano I., et al. Effetti del blocco cronico dei canali del calcio sull'attività del nervo simpatico nell'ipertensione. Ipertensione 2002; 39(4): 892-6.
61. Lefrandt J.D., Heitmann J., Sevre K., et al. Gli effetti delle classi di calcioantagonisti diidropiridina e fenilalchilammina sulla funzione autonomica nell'ipertensione: lo studio VAMPHYRE. Am J Hypertens 2001; 14(11): 1083-9.
62. Wanovich R., Kerrish P., Gerbino P.P., Shoheiber O. Modelli di compliance dei pazienti trattati con 2 agenti antipertensivi separati rispetto alla terapia di combinazione a dose fissa Am J Hypertens 2004; 175:223.
63. Dezii C.M. Uno studio retrospettivo sulla persistenza con la terapia combinata a pillola singola vs. terapia concomitante con due pillole in pazienti con ipertensione. Gestisci la cura. 2000; 9(9): 2-6.
64. Gerbino P.P., Shoheiber O. Modelli di aderenza tra i pazienti trattati con una combinazione a dose fissa rispetto ad agenti antipertensivi separati. Am J Health Syst Pharm 2007; 64(12): 1279-83.
65. Jackson K. Persistenza della combinazione fissa rispetto a quella libera con valsartan e HCTZ per pazienti con ipertensione. Valore fornitura sanitaria 2006; 9:363.
66. Aepfelbacher F.C., Messerli F.H., Nunez E., Michalewicz L. Effetti cardiovascolari di una combinazione trandolapril/verapamil in pazienti con ipertensione essenziale da lieve a moderata. Am J Cardiol 1997; 79(6): 826-8.
67. Reynolds N.A., Wagstaff A.J., Keam S.J. Trandolapril/verapamil a rilascio prolungato: una revisione del suo utilizzo nel trattamento dell'ipertensione essenziale. Farmaci 2005; 65(13): 1893-914.
68. Sharma S.K., Ruggenenti P., Remuzzi G. Gestione dell'ipertensione nei pazienti diabetici - focus sulla combinazione trandolapril/verapamil. Vasc Health Risk Management 2007; 3(4): 453-65.
69. Bakris G., Molitch M., Hewkin A., et al. Differenze nella tolleranza al glucosio tra combinazioni di farmaci antipertensivi a dose fissa nelle persone con sindrome metabolica. Cura del diabete 2006; 29(12): 2592-7.
70. Bakris G., Molitch M., Zhou Q., et al. Inversione della ridotta tolleranza al glucosio associata ai diuretici e del diabete di nuova insorgenza: risultati dello studio STAR-LET. Sindrome di J Cardiometab 2008; 3(1): 18-25.
71. Bailey C.J., Day C. Nuovi approcci farmacologici all'obesità. Pratica sull'obesità 2005; 1:2-5.
72. Yanovski S.Z., Yanovski J.A.Y. Obesità. N Engl J Med 2002; 346(8): 591-602.
73. Giorno C., Bailey C.J. Aggiornamento sulla sibutramina. Br J Diabet Vasc Dis 2002; 2: 392-7.
74. James W.P., Astrup A., Finer N., et al. Effetto della sibutramina sul mantenimento del peso dopo la perdita di peso: uno studio randomizzato. Gruppo di studio TEMPESTA. Prova di sibutramina per la riduzione e il mantenimento dell'obesità. Lancetta 2000; 356:2119-25.
75. Torp-Pedersen C., Caterson I., Coutinho W., et al. Risposte cardiovascolari al controllo del peso e alla sibutramina in soggetti ad alto rischio: un'analisi dello studio SCOUT. Eur Cuore J 2007; 28(23): 2915-23.
76. Sharma A.M., Caterson I.D., Coutinho W., et al. Cambiamenti della pressione arteriosa associati alla sibutramina e al controllo del peso: un'analisi del periodo iniziale di 6 settimane dello studio sugli esiti cardiovascolari della sibutramina (SCOUT). Diabete Obes Metab 2008. doi 10.1111/J.1463-1326.2008.00930.
77. Nakou E., Filippatos T.D., Liberopoulos E.N., et al. Effetti della combinazione sibutramina più verapamil a rilascio prolungato/trandolapril sulla pressione sanguigna e sulle variabili metaboliche nei pazienti ipertesi obesi. Opinione degli esperti Pharmacother 2008; 9(10): 1629-39.
78. Scholze J., Grimm E., Herrmann D., et al. Trattamento ottimale dell'ipertensione correlata all'obesità: lo studio Hypertension-Obesity-Sibutramine (HOS). Circolazione 2007; 115(15): 1991-8.
79.Glezer M.G. Risultati dello studio russo POLONESE (Efficacia e sicurezza di enarenal in pazienti con ipertensione arteriosa). Archivio Terapeutico 2006; 4: 44-50.
Per preventivo: Leonova M.V. Farmaci nuovi e promettenti che bloccano il sistema renina-angiotensina-aldosterone // Cancro al seno. Revisione medica. 2013. N. 17. P.886
Il ruolo del sistema renina-angiotensina-aldosterone (RAAS) nello sviluppo dell’ipertensione arteriosa (AH) e di altre malattie cardiovascolari è attualmente considerato dominante. Nel continuum cardiovascolare, l’ipertensione è tra i fattori di rischio e il principale meccanismo fisiopatologico di danno al sistema cardiovascolare è l’angiotensina II (ATII). L'ATII è un componente chiave del RAAS, un effettore che implementa la vasocostrizione, la ritenzione di sodio, l'attivazione del sistema nervoso simpatico, la proliferazione e l'ipertrofia cellulare, lo sviluppo dello stress ossidativo e l'infiammazione della parete vascolare.
Attualmente, due classi di farmaci che bloccano il RAAS sono già state sviluppate e ampiamente utilizzate clinicamente: gli ACE inibitori e i bloccanti dei recettori ATII. Gli effetti farmacologici e clinici di queste classi differiscono. L'ACE è una peptidasi del gruppo delle metalloproteinasi dello zinco che metabolizza ATI, AT1-7, bradichinina, sostanza P e molti altri peptidi. Il meccanismo d'azione degli ACE inibitori è principalmente associato alla prevenzione della formazione di ATII, che promuove la vasodilatazione, la natriuresi ed elimina gli effetti proinfiammatori, proliferativi e altri effetti dell'ATII. Inoltre, gli ACE inibitori prevengono la degradazione della bradichinina e ne aumentano i livelli. La bradichinina è un potente vasodilatatore, potenzia la natriuresi e, soprattutto, ha un effetto cardioprotettivo (previene l'ipertrofia, riduce il danno ischemico al miocardio, migliora l'afflusso di sangue coronarico) e effetti vasoprotettivi, migliorando la funzione endoteliale. Allo stesso tempo, un elevato livello di bradichinina è la causa dello sviluppo di angioedema, che è uno dei gravi svantaggi degli ACE inibitori, che aumentano significativamente il livello di chinina.
Non sempre gli ACE inibitori sono in grado di bloccare completamente la formazione di ATII nei tessuti. È ormai accertato che anche altri enzimi non correlati all'ACE possono partecipare alla sua trasformazione nei tessuti, in primis le endopeptidasi, che non sono influenzate dagli ACE inibitori. Di conseguenza, gli ACE inibitori non possono eliminare completamente gli effetti dell’ATII, il che potrebbe essere la ragione della loro mancanza di efficacia.
La soluzione a questo problema è stata facilitata dalla scoperta dei recettori ATII e della prima classe di farmaci che bloccano selettivamente i recettori AT1. Attraverso i recettori AT1 si realizzano gli effetti avversi dell'ATII: vasocostrizione, secrezione di aldosterone, vasopressina, norepinefrina, ritenzione di liquidi, proliferazione di cellule muscolari lisce e cardiomiociti, attivazione del SAS, nonché un meccanismo di feedback negativo - la formazione di renina . I recettori AT2 svolgono funzioni “utili”, come vasodilatazione, processi di riparazione e rigenerazione, effetti antiproliferativi, differenziazione e sviluppo dei tessuti embrionali. Gli effetti clinici dei bloccanti dei recettori ATII sono mediati dall'eliminazione degli effetti "dannosi" di ATII a livello dei recettori AT1, il che garantisce un blocco più completo degli effetti avversi di ATII e un aumento dell'influenza di ATII sui recettori AT2 , che completa gli effetti vasodilatatori e antiproliferativi. I bloccanti dei recettori ATII hanno un effetto specifico sul RAAS senza interferire con il sistema delle chinine. La mancanza di influenza sull'attività del sistema chinina, da un lato, riduce la gravità degli effetti indesiderati (tosse, angioedema), ma, dall'altro, priva i bloccanti dei recettori ATII di un importante effetto antiischemico e vasoprotettivo, che li distingue dagli ACE inibitori. Per questo motivo le indicazioni all’uso degli antagonisti dei recettori ATII ripetono per lo più le indicazioni all’uso degli ACE inibitori, rendendoli farmaci alternativi.
Nonostante l’introduzione dei bloccanti del RAAS nella pratica diffusa nel trattamento dell’ipertensione, permangono problemi relativi al miglioramento dei risultati e della prognosi. Questi includono: la possibilità di migliorare il controllo della pressione arteriosa nella popolazione, l’efficacia del trattamento dell’ipertensione resistente e la possibilità di ridurre ulteriormente il rischio di malattie cardiovascolari.
La ricerca di nuovi modi per influenzare il RAAS continua attivamente; Sono in fase di studio altri sistemi strettamente interagenti e si stanno creando farmaci con molteplici meccanismi d'azione, come gli inibitori degli ACE e dell'endopeptidasi neutra (NEP), gli inibitori dell'enzima di conversione dell'endotelina (ACE) e dei NEP, gli inibitori ACE/NEP/EGT.
Inibitori della vasopeptidasi
Oltre al noto ACE, le vasopeptidasi comprendono altre due metalloproteinasi dello zinco: la neprilisina (endopeptidasi neutra, NEP) e l'enzima di conversione dell'endotelina, che possono anche essere bersagli dell'azione farmacologica.
La neprilisina è un enzima prodotto dall'endotelio vascolare e coinvolto nella degradazione del peptide natriuretico e della bradichinina.
Il sistema del peptide natriuretico è rappresentato da tre diverse isoforme: peptide natriuretico atriale (tipo A), peptide natriuretico cerebrale (tipo B), che sono sintetizzati nell'atrio e nel miocardio, e peptide C endoteliale, che nelle loro funzioni biologiche sono inibitori endogeni del RAAS e dell'endotelina-1 (Tabella 1). Gli effetti cardiovascolari e renali del peptide natriuretico comprendono una diminuzione della pressione sanguigna attraverso il suo effetto sul tono vascolare e sull'equilibrio idroelettrolitico, nonché effetti antiproliferativi e antifibrotici sugli organi bersaglio. Le prove più recenti suggeriscono che il sistema dei peptidi natriuretici è coinvolto nella regolazione metabolica: ossidazione dei lipidi, formazione e differenziazione degli adipociti, attivazione dell’adiponectina, secrezione di insulina e tolleranza ai carboidrati, che possono fornire protezione contro lo sviluppo della sindrome metabolica.
È ormai noto che lo sviluppo di malattie cardiovascolari è associato alla disregolazione del sistema dei peptidi natriuretici. Pertanto, nell'ipertensione si verifica una carenza di peptide natriuretico, che porta a sensibilità al sale e ridotta natriuresi; nell'insufficienza cardiaca cronica (ICC), sullo sfondo di una carenza, si osserva un funzionamento anormale degli ormoni del sistema del peptide natriuretico.
Pertanto, per potenziare il sistema dei peptidi natriuretici al fine di ottenere ulteriori effetti cardiorenali ipotensivi e protettivi, è possibile utilizzare gli inibitori della NEP. L'inibizione della neprilisina porta al potenziamento degli effetti natriuretici, diuretici e vasodilatatori del peptide natriuretico endogeno e, di conseguenza, ad una diminuzione della pressione sanguigna. Tuttavia, la NEP è coinvolta anche nella degradazione di altri peptidi vasoattivi, in particolare ATI, ATII ed endotelina-1. Pertanto, il bilancio degli effetti degli inibitori della NEP sul tono vascolare è variabile e dipende dalla predominanza degli effetti costrittori e dilatatori. Con l'uso a lungo termine, l'effetto antipertensivo degli inibitori della neprilisina è debole a causa dell'attivazione compensatoria della formazione di ATII e di endotelina-1.
A questo proposito, la combinazione degli effetti degli ACE inibitori e degli inibitori NEP può potenziare significativamente gli effetti emodinamici e antiproliferativi come risultato di un meccanismo d’azione complementare, che ha portato alla creazione di farmaci con un duplice meccanismo d’azione, chiamati collettivamente vasopeptidasi. inibitori (Tabella 2, Fig. 1).
Gli inibitori noti della vasopeptidasi sono caratterizzati da vari gradi di selettività per NEP/ACE: omapatrilato - 8,9:0,5; fasidoprilato - 5.1:9.8; Sampatrilat - 8.0:1.2. Di conseguenza, gli inibitori della vasopeptidasi hanno acquisito un potenziale molto maggiore per ottenere un effetto ipotensivo, indipendentemente dall’attività del RAAS e dal livello di ritenzione di sodio, e nella protezione degli organi (regressione dell’ipertrofia, albuminuria, rigidità vascolare). Il più studiato negli studi clinici è stato l’omapatrilat, che ha mostrato una maggiore efficacia antipertensiva rispetto agli ACE inibitori, e nei pazienti con CHF ha portato ad un aumento della frazione di eiezione e a un miglioramento degli esiti clinici (studi IMPRESS, OVERTURE), ma senza vantaggi rispetto agli ACE inibitori.
Tuttavia, in ampi studi clinici condotti con omapatrilato, è stata riscontrata una maggiore incidenza di angioedema rispetto agli ACE inibitori. È noto che l'incidenza di angioedema durante l'uso di ACE inibitori varia dallo 0,1 allo 0,5% nella popolazione, di cui il 20% dei casi è pericoloso per la vita, il che è associato ad un aumento multiplo delle concentrazioni di bradichinina e dei suoi metaboliti. I risultati dell’ampio studio multicentrico OCTAVE (n = 25.302), progettato specificamente per studiare l’incidenza dell’angioedema, hanno mostrato che l’incidenza di questo effetto collaterale durante il trattamento con omapatrilato supera quella del gruppo enalapril: 2,17% contro 0,68% ( rischio relativo 3.4) . Ciò è stato spiegato dall’aumento dell’effetto sui livelli di chinina con l’inibizione sinergica di ACE e NEP, associata all’inibizione dell’aminopeptidasi P, che è coinvolta nella degradazione della bradichinina.
Un nuovo inibitore duplice della vasopeptidasi che blocca ACE/NEP è ilepatril, che ha un’affinità maggiore per ACE rispetto a NEP. Studiando gli effetti farmacodinamici dell'ilepatril sull'attività del RAAS e del peptide natriuretico in volontari sani, si è scoperto che il farmaco sopprime l'ACE in modo dose-dipendente (in dosi di 5 e 25 mg) e significativamente (più dell'88%) nel plasma sanguigno per una durata superiore a 48 ore, indipendentemente dalla sensibilità al sale. Allo stesso tempo, il farmaco ha aumentato significativamente l’attività della renina plasmatica entro 48 ore e ha ridotto i livelli di aldosterone. Questi risultati hanno mostrato una soppressione pronunciata e più duratura del RAAS rispetto all’ACE inibitore ramipril alla dose di 10 mg, che è stata spiegata dall’effetto tissutale più significativo di ilepatril sull’ACE e da una maggiore affinità per l’ACE, e da un grado comparabile di blocco del RAAS rispetto alla combinazione di 150 mg di irbesartan + 10 mg di ramipril. Contrariamente all'effetto sul RAAS, l'effetto dell'ilepatril sul peptide natriuretico si è manifestato con un aumento a breve termine del livello della sua escrezione nel periodo 4-8 ore dopo l'assunzione di una dose di 25 mg, il che indica un abbassamento e affinità più debole per NEP e lo distingue da omapatrilat. Inoltre, in termini di livello di escrezione di elettroliti, il farmaco non ha un effetto natriuretico aggiuntivo rispetto al ramipril o all'irbesartan, come fanno altri inibitori della vasopeptidasi. L'effetto ipotensivo massimo si sviluppa 6-12 ore dopo l'assunzione del farmaco e la diminuzione della pressione arteriosa media è di 5±5 e 10±4 mmHg. rispettivamente a bassa e alta sensibilità al sale. Secondo le caratteristiche farmacocinetiche, ilepatril è un profarmaco con un metabolita attivo, che si forma rapidamente, raggiungendo la concentrazione massima dopo 1-1,5 ore e viene lentamente eliminato. Sono attualmente in corso studi clinici di fase III.
Una via alternativa alla doppia soppressione di RAAS e NEP è rappresentata da una combinazione di blocco dei recettori ATII e NEP (Fig. 2). I bloccanti dei recettori ATII non influenzano il metabolismo delle chinine, a differenza degli ACE inibitori, e quindi potenzialmente hanno un rischio minore di sviluppare complicanze di angioedema. Attualmente, il primo farmaco, LCZ696, un bloccante del recettore ATII con l'effetto di inibire la NEP in un rapporto 1:1, è sottoposto a studi clinici di fase III. La molecola del farmaco combinato contiene valsartan e un inibitore della NEP (AHU377) sotto forma di profarmaco. In un ampio studio condotto su pazienti con ipertensione (n=1.328), LCZ696 a dosi di 200-400 mg ha mostrato un vantaggio nell'effetto ipotensivo rispetto a valsartan a dosi di 160-320 mg sotto forma di un'ulteriore riduzione della pressione sanguigna di 5 /3 e 6/3 mmHg . . L'effetto ipotensivo di LCZ696 è stato accompagnato da una diminuzione più pronunciata della pressione sanguigna del polso: di 2,25 e 3,32 mmHg. rispettivamente alle dosi di 200 e 400 mg, che è attualmente considerato un fattore prognostico positivo per l'effetto sulla rigidità della parete vascolare e sugli esiti cardiovascolari. Allo stesso tempo, uno studio sui biomarcatori neuroumorali durante il trattamento con LCZ696 ha mostrato un aumento del livello del peptide natriuretico con un grado di aumento comparabile del livello di renina e aldosterone rispetto a valsartan. La tollerabilità nei pazienti con ipertensione è stata buona e non sono stati segnalati casi di angioedema. Lo studio PARAMOUMT è stato ora completato su 685 pazienti con CHF e FE intatta. I risultati dello studio hanno mostrato che LCZ696 riduce più velocemente e in modo più significativo il livello di NT-proBNP (l'endpoint primario è un marcatore di maggiore attività del peptide natriuretico e prognosi sfavorevole nell'insufficienza cardiaca congestizia) rispetto a valsartan, e riduce anche le dimensioni del catetere sinistro atrio, che indica la regressione del suo rimodellamento. È attualmente in corso uno studio su pazienti con CHF e FE ridotta (studio PARADIGM-HF).
Inibitori del sistema endotelina
Il sistema endotelina svolge un ruolo importante nella regolazione del tono vascolare e del flusso sanguigno regionale. Tra le tre isoforme conosciute, l'endotelina-1 è la più attiva. Oltre ai noti effetti vasocostrittori, l'endotelina stimola la proliferazione e la sintesi della matrice intercellulare e, grazie al suo effetto diretto sul tono dei vasi renali, è coinvolta nella regolazione dell'omeostasi idroelettrolitica. Gli effetti dell'endotelina si realizzano attraverso l'interazione con specifici recettori di tipo A e di tipo B, le cui funzioni sono reciprocamente opposte: la vasocostrizione avviene attraverso i recettori di tipo A e la vasodilatazione avviene attraverso i recettori di tipo B. Negli ultimi anni è stato stabilito che i recettori di tipo B svolgono un ruolo importante nella clearance dell’endotelina-1, cioè quando questi recettori vengono bloccati, la clearance dell'endotelina-1 dipendente dal recettore viene interrotta e la sua concentrazione aumenta. Inoltre, i recettori di tipo B sono coinvolti nella regolazione degli effetti renali dell’endotelina-1 e nel mantenimento dell’omeostasi dei liquidi e degli elettroliti, che è importante.
Attualmente, il ruolo dell'endotelina è stato dimostrato nello sviluppo di una serie di malattie, incl. ipertensione, CHF, ipertensione polmonare, malattia renale cronica; è stata dimostrata una stretta relazione tra i livelli di endotelina e la sindrome metabolica, la disfunzione endoteliale e l'aterogenesi. Dagli anni '90 è in corso la ricerca di antagonisti dei recettori dell'endotelina adatti all'uso clinico; Sono già noti 10 farmaci (“sentans”) con vari gradi di selettività per i recettori di tipo A/B. Il primo antagonista non selettivo dei recettori dell’endotelina, il bosentan, in uno studio clinico condotto su pazienti con ipertensione ha mostrato un’efficacia antipertensiva paragonabile a quella dell’ACE inibitore enalapril. Ulteriori studi sull'efficacia dell'uso degli antagonisti dell'endotelina nell'ipertensione hanno mostrato il loro significato clinico nel trattamento dell'ipertensione resistente e dell'alto rischio cardiovascolare. Questi dati sono stati ottenuti da due ampi studi clinici, DORADO (n=379) e DORADO-AC (n=849), in cui darusentan è stato aggiunto alla terapia di tripla combinazione in pazienti con ipertensione resistente. Nello studio DORADO, nei pazienti con ipertensione resistente combinata con malattia renale cronica e proteinuria, a seguito dell'aggiunta di darusentan, è stata osservata non solo una significativa diminuzione della pressione sanguigna, ma anche una diminuzione dell'escrezione proteica. L'effetto antiproteinurico degli antagonisti dei recettori dell'endotelina è stato successivamente confermato in uno studio condotto su pazienti con nefropatia diabetica trattati con avosentan. Tuttavia, nello studio DORADO-AS non sono stati riscontrati vantaggi in termini di ulteriore riduzione della pressione arteriosa rispetto ai farmaci di confronto e al placebo, motivo per cui sono stati interrotti ulteriori studi. Inoltre, 4 ampi studi sugli antagonisti dell’endotelina (bosentan, darusentan, enrasentan) in pazienti con CHF hanno mostrato risultati contrastanti, spiegati da un aumento delle concentrazioni di endotelina-1. Ulteriori studi sugli antagonisti dei recettori dell'endotelina sono stati sospesi a causa degli effetti avversi associati alla ritenzione di liquidi (edema periferico, sovraccarico di volume). Lo sviluppo di questi effetti è associato all'effetto degli antagonisti dell'endotelina sui recettori di tipo B, che ha cambiato la ricerca di farmaci che influenzano il sistema endotelinico attraverso altre vie; e gli antagonisti dei recettori dell'endotelina hanno attualmente una sola indicazione: il trattamento dell'ipertensione polmonare.
Tenendo conto dell'elevata importanza del sistema endotelina nella regolazione del tono vascolare, è in corso la ricerca di un altro meccanismo d'azione attraverso la vasopeptidasi - EPF, che è coinvolto nella formazione dell'endotelina-1 attiva (Fig. 3). Il blocco dell’ACE e la combinazione con l’inibizione della NEP possono sopprimere efficacemente la formazione di endotelina-1 e potenziare gli effetti del peptide natriuretico. I vantaggi del duplice meccanismo d’azione sono, da un lato, quello di prevenire gli svantaggi degli inibitori NEP associati alla possibile vasocostrizione mediata dall’attivazione dell’endotelina, dall’altro, l’attività natriuretica degli inibitori NEP consente di compensare la ritenzione di liquidi associata alla blocco non selettivo dei recettori dell'endotelina. Daglutril è un doppio inibitore NEP e ACE in fase di sperimentazione clinica di fase II. Gli studi hanno dimostrato effetti cardioprotettivi pronunciati del farmaco dovuti alla diminuzione del rimodellamento cardiaco e vascolare, alla regressione dell'ipertrofia e della fibrosi.
Inibitori diretti della renina
È noto che gli ACE inibitori e i bloccanti dei recettori ATII aumentano l’attività della renina attraverso un meccanismo di feedback, motivo per cui l’efficacia dei bloccanti RAAS sfugge. La renina rappresenta il primo passo della cascata RAAS; è prodotto dalle cellule iuxtaglomerulari dei reni. La renina, attraverso l'angiotensinogeno, favorisce la formazione di ATII, la vasocostrizione e la secrezione di aldosterone, e regola anche i meccanismi di feedback. Pertanto, l’inibizione della renina ci consente di ottenere un blocco più completo del sistema RAAS. La ricerca sugli inibitori della renina è in corso dagli anni '70; Per molto tempo non è stato possibile ottenere una forma orale di inibitori della renina a causa della loro bassa biodisponibilità nel tratto gastrointestinale (meno del 2%). Il primo inibitore diretto della renina adatto per uso orale, aliskiren, è stato registrato nel 2007. Aliskiren ha una bassa biodisponibilità (2,6%), una lunga emivita (24-40 ore) e una via di eliminazione extrarenale. La farmacodinamica di aliskiren è associata ad una diminuzione dell'80% dei livelli di ATII. Negli studi clinici condotti su pazienti con ipertensione, aliskiren a dosi di 150-300 mg/die ha portato ad una diminuzione della pressione sistolica di 8,7-13 e 14,1-15,8 mmHg. rispettivamente e DBP - di 7,8-10,3 e 10,3-12,3 mm Hg. . L'effetto ipotensivo di aliskiren è stato osservato in diversi sottogruppi di pazienti, inclusi pazienti con sindrome metabolica, obesità; in termini di gravità l'effetto è stato paragonabile a quello degli ACE inibitori e dei bloccanti dei recettori ATII ed è stato osservato un effetto additivo anche in combinazione con valsartan, idroclorotiazide e amlodipina. Numerosi studi clinici hanno dimostrato gli effetti organoprotettivi del farmaco: effetto antiproteinurico in pazienti con nefropatia diabetica (studio AVOID, n=599), regressione dell'ipertrofia ventricolare sinistra in pazienti con ipertensione (studio ALLAY, n=465). Pertanto, nello studio AVOID, dopo 3 mesi di trattamento con losartan alla dose di 100 mg/die e al raggiungimento del livello di pressione arteriosa target (<130/80 мм рт.ст.) при компенсированном уровне гликемии (гликированный гемоглобин 8%) больных рандомизировали к приему алискирена в дозах 150-300 мг/сут или плацебо. Отмечено достоверное снижение индекса альбумин/креатинин в моче (первичная конечная точка) на 11% через 3 мес. и на 20% - через 6 мес. в сравнении с группой плацебо. В ночное время экскреция альбумина на фоне приема алискирена снизилась на 18%, а доля пациентов со снижением экскреции альбумина на 50% и более была вдвое большей (24,7% пациентов в группе алискирена против 12,5% в группе плацебо) . Причем нефропротективный эффект алискирена не был связан со снижением АД. Одним из объяснений выявленного нефропротективного эффекта у алискирена авторы считают полученные ранее в экспериментальных исследованиях на моделях диабета данные о способности препарата снижать количество рениновых и прорениновых рецепторов в почках, а также уменьшать профибротические процессы и апоптоз подоцитов, что обеспечивает более выраженный эффект в сравнении с эффектом ингибиторов АПФ . В исследовании ALLAY у пациентов с АГ и увеличением толщины миокарда ЛЖ (более 1,3 см по данным ЭхоКГ) применение алискирена ассоциировалось с одинаковой степенью регресса ИММЛЖ в сравнении с лозартаном и комбинацией алискирена с лозартаном: −5,7±10,6 , −5,4±10,8, −7,9±9,6 г/м2 соответственно. У части пациентов (n=136) проводилось изучение динамики нейрогормонов РААС, и было выявлено достоверное и значительное снижение уровня альдостерона и активности ренина плазмы на фоне применения алискирена или комбинации алискирена с лозартаном, тогда как на фоне применения монотерапии лозартаном эффект влияния на альдостерон отсутствовал, а на активность ренина - был противоположным, что объясняет значимость подавления альдостерона в достижении регресса ГЛЖ.
Inoltre, sono in corso una serie di studi clinici sull'aliskiren nel trattamento di altre malattie cardiovascolari per valutare l'effetto sulla prognosi dei pazienti: ALOFT (n=320), ASTRONAUT (n=1639), ATMOSPHERE (n=7000 ) studi in pazienti con CHF, lo studio ALTITUDE in pazienti con diabete mellito e ad alto rischio cardiovascolare, lo studio ASPIRE in pazienti con rimodellamento post-infarto.
Conclusione
Per risolvere i problemi di prevenzione delle malattie cardiovascolari, continua la creazione di nuovi farmaci con un complesso meccanismo d'azione multiplo, che consentano un blocco più completo del RAAS attraverso una cascata di meccanismi di regolazione emodinamica e neuroumorale. I potenziali effetti di tali farmaci consentono non solo di fornire un ulteriore effetto ipotensivo, ma anche di ottenere il controllo della pressione arteriosa nei pazienti ad alto rischio, compresa l'ipertensione resistente. I farmaci con molteplici meccanismi d'azione presentano vantaggi in un effetto organoprotettivo più pronunciato, che previene ulteriori danni al sistema cardiovascolare. Lo studio dei benefici dei nuovi farmaci che bloccano il RAAS richiede ulteriori ricerche e valutazioni del loro impatto sulla prognosi dei pazienti con ipertensione e altre malattie cardiovascolari.


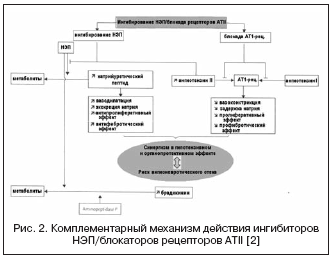
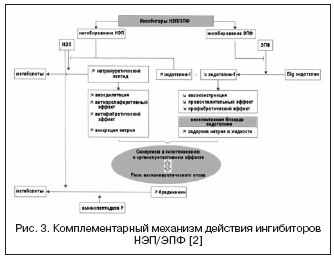
Letteratura
1. Campbell D.J. Inibizione della vasopeptidasi: un’arma a doppio taglio? // Ipertensione. 2003.vol. 41. P. 383-389.
2. Laurent S., Schlaich M., Esler M. Nuovi farmaci, procedure e dispositivi per l'ipertensione // Lancet. 2012. vol. 380. P. 591-600.
3. Corti R., Burnett J.C., Rouleau J.L. et al. Inibitori della vasopeptidasi: un nuovo concetto terapeutico nelle malattie cardiovascolari? // Circolazione. 2001.vol. 104. P. 1856-1862.
4. Mangiafico S., Costello-Boerrigter L.C., Andersen I.A. et al. Inibizione dell'endopeptidasi neutra e sistema del peptide natriuretico: una strategia in evoluzione nella terapia cardiovascolare // Eur. Cuore J. 2012, doi:10.1093/eurheartj/ehs262.
5. Rouleau J.L., Pfeffer M.A., Stewart D.J. et al. Confronto tra inibitore della vasopeptidasi, omapatrilato e lisinopril sulla tolleranza all'esercizio e sulla morbilità nei pazienti con insufficienza cardiaca: studio randomizzato IMPRESS // Lancet. 2000.vol. 356. P. 615-620.
6. Packer M., Califf R.M., Konstam M.A. et al. Confronto tra omapatrilat ed enalapril in pazienti con insufficienza cardiaca cronica: studio randomizzato di utilità nella riduzione degli eventi di Omapatrilat rispetto a enalapril (OVERTURE) // Circolazione. 2002.vol. 106. P. 920-926.
7. Warner K.K., Visconti J.A., Tschampel M.M. Bloccanti dei recettori dell'angiotensina II in pazienti con angioedema indotto da ACE inibitori // Ann. Farmacoterapista. 2000.vol. 34. P. 526-528.
8. Kostis J.B., Packer M., Black H.R. et al. Omapatrilat ed enalapril in pazienti con ipertensione: lo studio Omapatrilat Cardiovascolare vs Enalapril (OCTAVE) // Am. J. ipertenico. 2004.vol. 17. P. 103-111.
9. Azizi M., Bissery A., Peyrard S. et al. Farmacocinetica e farmacodinamica dell'inibitore della vasopeptidasi AVE7688 nell'uomo // Clin. Farmaco. Là. 2006.vol. 79. P. 49-61.
10. Gu J., Noe A., Chandra P. et al. Farmacocinetica e farmacodinamica di LCZ696, un nuovo inibitore della neprilisina del recettore dell'angiotensina a doppia azione (ARNi) // J. Clin. Farmaco. 2010.vol. 50. P. 401-414.
11. Ruilope L.M., Dukat A., Buhm M. et al. Riduzione della pressione arteriosa con LCZ696, un nuovo inibitore a doppia azione del recettore dell'angiotensina II e della neprilisina: uno studio di confronto attivo randomizzato, in doppio cieco, controllato con placebo // Lancet. 2010.vol. 375. P. 1255-1266.
12. Solomon S.D., Zile M., Pieske B. et al. L'inibitore della neprilisina del recettore dell'angiotensina LCZ696 nell'insufficienza cardiaca con frazione di eiezione preservata: uno studio controllato randomizzato in doppio cieco di fase 2 // Lancet. 2012. vol. 380(9851). P. 1387-1395.
13. Levin E.R. Endoteline // N. Engl. J.Med. 1995.vol. 333. P. 356-363.
14. Dhaun N., Goddard J., Kohan D.E. et al. Ruolo dell'endotelina-1 nell'ipertensione clinica: 20 anni dopo // Ipertensione. 2008.vol. 52. P. 452-459.
15. Burnier M., Forni V. Antagonisti dei recettori dell'endotelina: un posto nella gestione dell'ipertensione essenziale? // Nefrolo. Comporre. Trapianto. 2011. 0: 1-4. doi: 10.1093/ndt/gfr704.
16. Krum H., Viskoper R. J., Lacourciere Y. et al. L'effetto di un antagonista del recettore dell'endotelina, bosentan, sulla pressione sanguigna in pazienti con ipertensione essenziale. Investigatori sull'ipertensione Bosentan // N. Engl. J.Med. 1998.vol. 338. P. 784-790.
17. Weber M.A., Black H., Bakris G. et al. Un antagonista selettivo del recettore dell'endotelina per ridurre la pressione sanguigna nei pazienti con ipertensione resistente al trattamento: uno studio randomizzato, in doppio cieco, controllato con placebo // Lancet. 2009.vol. 374. P. 1423-1431.
18. Bakris G.L., Lindholm L.H., Black H.R. et al. Risultati divergenti utilizzando la pressione sanguigna clinica e ambulatoriale: rapporto di uno studio sull'ipertensione resistente a darusentan // Ipertensione. 2010.vol. 56. P. 824-830.
19. Mann J.F., Green D., Jamerson K. et al. Avosentan per la nefropatia diabetica conclamata // J. Am. Soc. Nefrolo. 2010.vol. 21. P. 527-535.
20. Kalk P., Sharkovska Y., Kashina E. et al. L'enzima di conversione dell'endotelina/inibitore neutro dell'endopeptidasi SLV338 previene il rimodellamento cardiaco ipertensivo in modo indipendente dalla pressione sanguigna // Ipertensione. 2011. vol. 57. P. 755-763.
21. Nussberger J., Wuerzner G., Jensen C. et al. Soppressione dell'angiotensina II nell'uomo mediante l'inibitore della renina teoricamente attivo Aliskiren (SPP100): confronto con enalapril // Ipertensione. 2002.vol. 39(1). P.E1-8.
22. Alreja G., Joseph J. Renin e le malattie cardiovascolari: percorso di logoramento o nuova direzione? // Mondo J. Cardiol. 2011. vol. 3(3). P.72-83.
23. Ingelfinger J.R. Aliskiren e duplice terapia nel diabete mellito di tipo 2 // N. Engl. J.Med. 2008.vol. 358(23). P.2503-2505.
24. Pouleur A.S., Uno H., Prescott M.F., Desai A. (per gli investigatori ALLAY). La soppressione dell'aldosterone media la regressione dell'ipertrofia ventricolare sinistra nei pazienti con ipertensione // J. Sistema renina-angiotensina-aldosterone. 2011. vol. 12. P. 483-490.
25. Kelly D.J., Zhang Y., Moe G. et al. Aliskiren, un nuovo inibitore della renina, è renoprotettivo in un modello di nefropatia diabetica avanzata nei ratti // Diabetol. 2007.vol. 50. P. 2398-2404.