Синдром на Олбрайт - рядко генетично заболяванес неизвестен произход. Унаследява се и се характеризира предимно с увреждане скелетна система. Най-често се среща при жени. Но има моменти, когато момчетата също могат да се разболеят. Повечето пациенти с този синдром страдат от физическо и умствено изоставане.
Болестта е кръстена на учения, който я е открил. Скелетната система започва да се разрушава под въздействието на метаболизма на калция и фосфора. Паращитовидната жлеза отделя паратироиден хормон; при човек със синдром на Олбрайт тъканната резистентност към паратироидния хормон е нарушена.
Симптоми на синдрома на Бътлър Олбрайт
Има три основни характеристикизаболявания. Ако са налице дори две от три, може да се постави такава диагноза.
- Много ранен пубертет в резултат на силно повишени хормонални нива. Момичетата могат да започнат менструация през първите месеци след раждането. Момчетата със синдром на Олбрайт много рядко изпитват ранен пубертет. Промени в хормонален фонвъзниква поради неправилно функциониране на надбъбречните жлези, хипофизата и щитовидната жлеза. Този симптом може да се появи в пълна или непълна форма. При пълна форма менструацията започва много рано и обикновено е обилна и болезнена. В ранна детска възраст този синдром проявява и вторични сексуални характеристики, например подуване на млечните жлези. Ако изявата е непълна, менструацията не настъпва. Много често със синдрома на Олбрайт момичетата развиват кисти на яйчниците. При момчетата по време на ранния пубертет се наблюдава уголемяване на пениса и окосмяване на пубиса.
- Фиброзна остеодисплазия. Това заболяване показва нарушение на костната тъкан, което води до изкривяване и деформация на костите. Характеризира се с асиметрия. Най-честите признаци са куцота и изкривяване на гръбначния стълб. Най-често от промени са засегнати дългите кости. Растежът на скелета се забавя, това се забелязва дори при деца. Много жертви на синдрома на McCune изпитват неравномерен растеж на костите на черепа, което води до външна деформация. Когато лицето се промени, може да се появят изпъкнали очи. След навършване на пубертета промени в коститезабавят и честотата на фрактурите намалява.
- Промени в кожата. Проявява се в повишена пигментация. Кожата на торса и бедрата се покрива с жълто-кафяви петна с назъбени ръбове. Пигментация може да се види в долната част на гърба, горната част на гърба, задните части и гърдите. Петната се появяват веднага след раждането на бебето.

Лечение на синдрома на Олбрайт (McCune).
 Пациенти, страдащи от синдром на McCune, трябва през цялото време да бъде под наблюдението на специализирани специалисти (гинеколог, ендокринолог, травматолог, офталмолог).
Пациенти, страдащи от синдром на McCune, трябва през цялото време да бъде под наблюдението на специализирани специалисти (гинеколог, ендокринолог, травматолог, офталмолог).
Необходимо комплексно лечение. Тъй като заболяването се характеризира генетична мутация, тогава е необходимо преди всичко да се отървете от симптомите на неговото проявление.
Показано е задължително хормонално лечение за поддържане на ендокринната система.
Необходимо е да не пропускате момента, в който костите на лицето започват да се деформират, за да предотвратите тези промени по време на заболяване.
Понякога се налага хирургична интервенция . Предписва се, когато поради промени в структурата на черепа съществува риск от загуба на слуха или зрението.
При болка се използват специални болкоуспокояващи, съдържащи малки количества бифосфонати.
Много е важно да се извършва предпазни меркиза укрепване на мускулите.
За всеки пациент, страдащ от болестта на Олбрайт, лекарят избира индивидуално лечение, което трябва стриктно да се спазва.
Трябва да се наблюдава постоянноколичество калций в тялото. При започване на терапията такива прегледи се провеждат веднъж седмично, след което честотата им се намалява до веднъж месечно до приключване на основното лечение.
По време на лечението е много важно да се спазва диета, при която количеството храни, съдържащи фосфор, е намалено до минимум.
Трябва да бъде отбелязано че, жена, страдаща от този синдромКогато решавате да забременеете, определено трябва да се консултирате с добър генетик.
Болестта на Олбрайт (McCune) се среща при един човек на 1 000 000. Специфично лечение за този синдром все още не е изобретено, но човек не трябва да губи надежда. Медицината не стои неподвижна и лекарите ще помогнат за преодоляване на неприятните моменти и ще държат пациента под наблюдение през цялото време.
Гените са източникът на цялата информация за структурата и функционирането на тялото. От тяхното правилно функциониране зависи буквално всичко: образуването на органи и тъкани, възстановяването на увредените структури и образуването на нови, както и обмяната на веществата. Всички химически трансформации в тялото са записани в гените. Метаболизмът се осъществява с помощта на специални ускорители - ензими. Тяхната неправилна структура води до много проблеми. Подобни наследствени патологии включват болестта на Олбрайт.
Обмяна на калций и фосфор в организма
Метаболизмът е най-важният аспект от живота на човешкото тяло. Всяка секунда само в тъкани и органи химически елементипревръщат се в други. По правило тяхното съдържание е строго регламентирано. Това важи особено за електролитите – натрий, калий, калций и фосфор.Благодарение на техните специфични взаимоотношения е възможна работата на сърцето и скелетните мускули и предаването на нервен електрически сигнал.
Калцият се намира във всички органи и тъкани и играе няколко важни роли:
- е строителен материал за костите;
Калцият е основният градивен елемент на костната тъкан
- осигурява функционирането на сърцето и скелетните мускули;
Калцият е един от участниците в мускулната контракция
- участва в кръвосъсирването и метаболизма;
- е посредник на действието на хормоните - специални химични сигнали, които регулират функционирането на тялото.
Фосфорът се намира в големи количества в костната тъкан и е от съществено значение за нормалната мозъчна функция. Калцият и фосфорът присъстват в кръвта в строго определено съотношение. Излишъкът от тези химични съединения се отделя от бъбреците с урината.
Паратироидният хормон е основният регулатор на нивата на калций и фосфор в кръвта
Благосъстоянието на тялото пряко зависи от постоянното количество калций и фосфор. Процесът на тяхната обмяна се контролира от щитовидната и паращитовидните жлези:
- Първият произвежда хормона калцитонин, благодарение на който калцият се прехвърля от кръвта в костната тъкан.
- Последните, използвайки паратиреоиден хормон, премахват липсата на калций в кръвта, като го измиват от костите.
В допълнение, обмяната на калций и фосфор се регулира от бъбреците, премахвайки излишъка от тези химични съединения в урината. Витамин D играе важна роля в това.
Синдромът на Олбрайт е наследствено заболяване, причинено от дефект в метаболизма на калция и фосфора в организма. Води до различни промени в костите, бъбреците, храносмилателния тракт и психични разстройства.
Синоними на болестта на Олбрайт: псевдохипопаратироидизъм, наследствена остеодистрофия.
Наследствената остеодистрофия е разгледана за първи път от учения Фулър Олбрайт през 1942 г. Заболяването се среща няколко пъти по-често при жените. IN медицинска литератураОписани са само около 300 случая на тази рядка патология.
Класификация
Псевдохипопаратироидизмът се разделя на три основни типа:

Причини и фактори за развитие
Псевдохипопаратиреоидизмът се основава на наследствената устойчивост на телесните тъкани към влиянието на паратироидния хормон. При нормални условия последният действа чрез специален посредник - цикличен аденозин монофосфат (цАМФ). Той е този, който инструктира клетката да се подчини на регулаторния сигнал на паратироидния хормон. При болестта на Олбрайт структурата на медиатора се променя на генетично ниво.
Болестта на Олбрайт се предава на следващите поколения по интересен модел, който се нарича Х-свързано доминантно унаследяване: патологията преминава от майката към дъщерите и синовете в 50% от случаите. Със същата вероятност само дъщерите наследяват дефектния ген от баща си. Има фамилни случаи на заболяването.
Генът на синдрома на Олбрайт се намира на половата хромозома
Без регулаторното действие на паратироидния хормон метаболизмът на калция се измества значително към използване в костната тъкан. В този случай има негов дефицит в кръвта, което води до следните явления:
- мускулни крампи;
- нарушаване на образуването на електрически импулси в миокарда и промени в сърдечния ритъм;
- повишено производство на паратироиден хормон и уголемяване на паращитовидните жлези.
Излишъкът от паратироиден хормон, от своя страна, води до повишаване на нивата на фосфор в кръвта.
Дефектен ензим при болестта на Олбрайт нарушава дейността не само на паратироидния хормон. Патологията въвлича в своя порочен кръг хормоните на щитовидната и половите жлези. В някои случаи настъпва ранен пубертет - пубертет - поради високата активност на половите хормони.
Щитовидната и паращитовидните жлези са основните регулатори на метаболизма на калция и фосфора в организма.
Болест на Олбрайт - видео
Признаци на болестта на Олбрайт
Наследственото заболяване най-често засяга цялото тяло, което води до множество промени. Признаците на патология в този случай са изключително сходни с тези на хипопаратироидизма - дефицит на производство на паратироиден хормон в паращитовидните жлези.
Често срещан симптом на псевдохипопаратироидизма са мускулните крампи. Тяхната тежест зависи пряко от нивото на калций в кръвта: от потрепване на отделни влакна до пълен спазъм на целия скелетен мускул - опистотонус.
Симптоми на псевдохипопаратироидизъм - табл
| Локализация на промените | Скелет | Скелетни мускули | Сърдечен мускул | Бъбреци | Мозък и сетивни органи | Кожа, мастна и съединителна тъкан |
| Признаци на заболяването |
|
|
|
|
| Образуване на подкожни натрупвания на калциеви кристали |
Диагностични методи
За разпознаване на болестта на Олбрайт е необходима съвместната работа на няколко специалисти: уролог, невролог, психиатър, ендокринолог, генетик. За установяване на правилна диагноза се използват следните методи:
- обективен преглед - ви позволява да идентифицирате нисък ръст, аномалии в структурата на ръцете и зъбите;
Болестта на Олбрайт причинява скъсяване на пръстите
- неврологичен преглед - помага за откриване на признаци на повишена конвулсивна готовност под формата на потрепване на отделни мускули на лицето, торса и крайниците;
- преглед от психиатър - позволява да се установи умствена изостаналост, интелектуални и паметови увреждания;
- биохимичен кръвен тест - ви позволява да откриете ниски нива на калций, високи нива на фосфор и нормални или повишени нива на паратироидния хормон;
Калцият и фосфорът са едни от основните елементи на течната част на кръвта
- биохимичен тест на урината - открива ниски нива на калций и фосфор;
- тест с въвеждането на паратиреоиден хормон - ви позволява да разграничите псевдохипопаратироидизма от истинския дефицит на паратироидния хормон. При болестта на Олбрайт въвеждането на паратиреоиден хормон в кръвта не предизвиква повишаване на количеството фосфор в урината;
- Рентгеново измерване на костната плътност (денситометрия) - диагностицира аномалии в структурата на ръката, области на размекване и изтъняване на костната тъкан, както и натрупване на калций в мускулите, кожата и подкожната тъкан;
При болестта на Олбрайт се променя плътността на костното вещество (вляво - нормално, вдясно - патология)
- компютърно (или магнитно резонансно) изображение на мозъка - ви позволява да идентифицирате огнища на натрупване на калций в отделни структури, например базалните ганглии, които са отговорни за координацията на движенията;
Калцият често се отлага в областта на базалните ганглии
- Ултразвуково изследване на бъбреците - помага да се установи наличието на камъни, техния брой и размер;
- Ултразвукът на паращитовидните жлези може да разкрие увеличаване на размера на органа, както и наличието на кисти.
Диференциална диагнозаизвършва се със следните заболявания:

Методи за лечение
Псевдохипопаратиреоидизмът изисква наблюдение през целия живот от ендокринолог. При повторни тежки пристъпи на конвулсии е необходима хоспитализация в специализирано болнично отделение.
Лекарствена терапия
Лечението с лекарства е основният компонент на терапията на псевдохипопаратироидизма. Основните цели са:
- нормализиране на нивата на калций и фосфор в кръвта;
- възстановяване на нормалната костна структура;
- предотвратяване на гърчове;
- предотвратяване образуването на камъни в бъбреците.
За нормализиране на нивата на калций се използват лекарствата Калциев хлорид и Калциев глюконат. Най-често тези лекарства се използват под формата на таблетки. Въпреки това, за премахване на мускулни крампи се практикува венозно приложениефармакологични средства.
Калциевият глюконат е лекарство, използвано за лечение на болестта на Олбрайт
Добавките с витамин D помагат за нормализиране на нивата на калций и фосфор в кръвта:
- Оксидевит;
- Калцитрин.
Витамин D също помага за укрепване на костната тъкан и зъбите.
Калций D3 се предписва при болестта на Олбрайт
Прилагането на паратироиден хормон при псевдохипопаратироидизъм е неефективно, тъй като тъканите и органите са нечувствителни към неговото влияние.
Корекция на диетата
Важен компонент от лечението на псевдохипопаратироидизъм е корекцията на диетата. В същото време достатъчен калориен прием и частични хранения са жизненоважни. Количеството течност, което пиете на ден, трябва да бъде най-малко 1,5-2 литра.
- боб;
- зелен грах;
- леща за готвене;
- бадем;
- кресон;
- целина;
- карфиол;
- копър;
- босилек;
- мляко;
- кисело мляко;
- сметана.
Храни с високо съдържание на калций - фотогалерия
 Фасулът е с високо съдържание на калций
Фасулът е с високо съдържание на калций  Соята е шампион сред бобовите растения по съдържание на калций (277 mg на 100 g)
Соята е шампион сред бобовите растения по съдържание на калций (277 mg на 100 g)  Кресонът е зеленина с високо съдържание на калций (81 mg на 100 g)
Кресонът е зеленина с високо съдържание на калций (81 mg на 100 g)  Целината се използва за приготвяне на зеленчукови салати
Целината се използва за приготвяне на зеленчукови салати  Босилекът се препоръчва да се използва като подправка
Босилекът се препоръчва да се използва като подправка  Ферментиралите млечни продукти съдържат много калций
Ферментиралите млечни продукти съдържат много калций
Храни с високо съдържание на фосфор, чиято консумация трябва да бъде ограничена:
- сусам;
- морска риба;
- яйчен жълтък;
- бадем;
- Шам-фъстъци;
- Кедрови ядки;
- фъстък;
- Орех.
Продукти с високо съдържание на фосфор - фотогалерия
 Морска риба - източник голямо количествофосфор
Морска риба - източник голямо количествофосфор  Консумацията на яйчен жълтък при патология трябва да се ограничи, а при болест на Олбрайт е по-добре да се въздържате от нахут.
Консумацията на яйчен жълтък при патология трябва да се ограничи, а при болест на Олбрайт е по-добре да се въздържате от нахут.  Орехиразличават високо нивофосфор
Орехиразличават високо нивофосфор
хирургия
Няма хирургично лечение на псевдохипопаратироидизъм. Помощта на хирурга е необходима при някои състояния, съпътстващи хода на заболяването:

Физиотерапия
Физиотерапевтичните мерки се използват при лечението на някои състояния, придружаващи хода на болестта на Олбрайт:
- с възпаление на чашките и таза на бъбреците (пиелонефрит) на фона на наличието на камъни;
- с възпаление на огнища на натрупване на калций в мускулите и подкожната мастна тъкан;
- за зарастване на фрактури.
За да се премахнат тези негативни явления, се използват следните методи:

Усложнения и прогноза
Прогнозата на терапията с навременно разпознаване на болестта на Олбрайт е относително благоприятна, но е невъзможно напълно да се излекува наследственият ензимен дефект. Нормализирането на съдържанието на калций и фосфор в организма ще позволи на пациентите да се занимават с лека работа, с изключение на нервно напрежениеи работа с машини и механизми.
В случай на повтарящи се пристъпи на мускулни крампи, които трудно се коригират с лекарства, медицинската комисия взема решение за определяне на група инвалидност. При тежки случаи на псевдохипопаратироидизъм могат да се развият следните усложнения:

Предотвратяване
Основен превантивна мярказа псевдохипопаратироидизъм е медицинско генетично консултиране. Генетик ще проучи родословието на съпрузите и ще определи вероятността от патология в потомството.
Псевдохипопаратироидизмът е тежко наследствено заболяване, включващо патологичен процесвсички тъкани и органи. Навременната консултация с лекар и спазването на препоръките за лечение ще позволи на пациента да запази работоспособността и качеството на живот.
За първи път в Руска литературатози синдром е описан от В. Р. Брайцев през 1922 г., определяйки го като "фиброзни тумори" 15 години по-късно ендокринологът Фулър Олбрайт формулира характеристиките на това системно заболяване като „синдром, характеризиращ се с дисеминиран фиброзен остеит, пигментирани полета и ендокринни нарушения с преждевременен пубертет при момичетата“.
С други думи, синдромът на Маккюн-Олбрайт-Брайцев е системно заболяване, характеризиращ се с преждевременно полово развитие и костна патология. Това заболяване се среща предимно при жените и е вродена наследствена патология. Доскоро се смяташе, че това е изключително женско заболяване, но след това бяха регистрирани случаи на заболяването и сред момчетата. Причината за синдрома се счита за мутация в специфичен ген.
Признаци на синдрома на McCune-Albright-Braitsev
- Тъмни петна.Всички пациенти имат специална пигментация с неправилна форма, цвят на кафе с мляко. Петната обикновено са разположени по гърба, гърдите, бедрата, задните части и долната част на гърба. Това е един вид маркер на заболяването, пигментацията присъства от раждането или се появява, когато детето расте.
- Костна патология.Дългите кости (като бедрената кост) обикновено са засегнати. Наличието на кухини в костите води до тяхната деформация, фрактури и огъване на крайниците. Типична деформация на тазобедрената става е типът „овчарски мошеник“. Възможно увреждане на костите на черепа. В този случай се появява асиметрия на лицето и едностранен екзофталм (изпъкнали очи). Уврежданията на костите и честотата на фрактурите намаляват след пубертета.
- Преждевременно сексуално развитие.Може да започне от 6-9 месечна възраст до 7 години. Най-често първите признаци на пубертета се появяват при дете на 3 години. Може да се прояви в пълна и непълна форма. Пълна формапри момичетата се характеризира с менархе (първа менструация) и уголемяване на млечните жлези. Дългосрочно и силно кървене, често се откриват кисти по яйчниците. При непълна форма менархе не настъпва.
Преждевременният пубертет при момчетата е много по-рядко срещан и е съпроводен със симетрично уголемяване на тестисите, след това растеж на пениса и пубисно окосмяване, както при нормалното полово развитие.
- Ускоряване на костната възраст и ускорено затваряне на растежните пластини.
- Неврологични и психични разстройства(умствена изостаналост, конвулсии, атрофия оптичен нерв, загуба на слуха).
- В литературата има информация за други ендокринни нарушения при това заболяване: акромегалия, хиперпаратироидизъм (повишена функция на паращитовидните жлези), хипертиреоидизъм (повишена функция на щитовидната жлеза) и наличие на възли в щитовидната жлеза, хиперкортицизъм (излишък на глюкокортикоидни хормони на надбъбречните жлези), рахит, устойчив на витамин D.
Диагностика на синдрома на McCune-Albright-Braitsev
- Наличие на характерни черти.
- Генетична консултация.
- рентгеново изследване.
- Полови хормони и гонадотропини, хормони на щитовидната жлеза, пролактин, други тропични хормони на хипофизата (по показания).
- Ултразвук на тазови органи за момичета и скротум за момчета.
- Консултация с ендокринолог и детски гинеколог за момичета или детски уролог за момчета.
- Изследване на други органи на ендокринната система (по показания).
Лечение на синдрома на McCune-Albright-Braitsev
Пациентите с тази патология се нуждаят от наблюдение на гинеколог-ендокринолог, ендокринолог, лекар радиологична диагностика, офталмолог, ортопед-травматолог. Освен това е необходимо периодично да се определя нивото на тропиците и гонадотропни хормони. Специфично лечениетози синдром не е разработен.Най-често лекарите избират тактика за наблюдение. За момичета терапията е показана в случай на продължително повишаване на нивата на естроген, придружено от чести и тежки кръвоизливи. По показания се извършва корекция на костни деформации. Когато се развият симптоми на увреждане на другите ендокринни жлезилечението се извършва от ендокринолози.
Наследствената остеодистрофия на Олбрайт (синоними: псевдохипопаратироидизъм тип 1а, болест на Олбрайт, синдром на Олбрайт) е рядко заболяване на скелетната система, причинено от мутация на ген, разположен на хромозома 20. Патологията се характеризира с нарушение на метаболизма на калция и фосфора, което засяга забавянето на физическото, а понякога и на умственото развитие. Начинът на наследяване на синдрома на Олбрайт все още се обсъжда.
За информация: американският ендокринолог Ф. Олбрайт е първият, който описва заболяването през 1942 г. В момента честотата на патологията е 79 случая на 10 милиона души. Жените боледуват два пъти по-често от мъжете.
Псевдохипопаратироидизъм: описание и симптоми
Заболяването наподобява хипопаратиреоидизъм - дефицит на паратхормон (паратиреоиден хормон). Механизмът на възникване обаче се различава по това, че няма дефицит на хормона, а генетичен дефект в комплекса от клетъчни рецептори в тъканите на бъбреците и скелета блокира действието му. Метаболитните процеси, които обикновено се регулират от паратироидния хормон чрез синтеза на сАМР, стават невъзможни. Паращитовидните жлези могат да бъдат компенсаторно увеличени.

В зависимост от вида на заболяването (и сега има четири клинични форми: типове 1 и 2, първият също е разделен на подтипове A, B и C), възможни са различни варианти за блокиране на действието на паратироидния хормон, както и нарушена резистентност към други хормони: глюкагон, TSH, GnRH, ADH. В резултат на това се развиват явления на хипотиреоидизъм, аменорея и се нарушава концентрационната функция на бъбреците.
Количеството фосфор в кръвта се увеличава, а нивото на активната форма на витамин D3 и калций намалява.
Външен вид на пациенти със синдром на Олбрайт
Заболяването води до множество аномалии в развитието на скелета, които се откриват при детство, понякога след година, но обикновено на 5-10 години и дори по-късно. Можете да подозирате болестта на Олбрайт при дете въз основа на следните признаци:
- лице с форма на луна;
- крилообразни гънки на шията;
- нисък ръст поради скъсени крака;
- затлъстяване;
- скъсяване на пръстите на ръцете и краката (брахидактилия);
- аплазия (недоразвитие) на зъбите;
- чести фрактури на костите;
- преждевременно сексуално развитие:
- множество подкожни области на калцификация.

важно! Всеки от тези симптоми изисква контакт с ендокринолог, предписване диагностични меркиза идентифициране на причините за състоянието и избор на адекватна терапия. От 90-те години на миналия век диагностиката се извършва на базата на молекулярно-генетични методи.
Ненавременният контакт с ендокринолог и генетик забавя правилната диагноза и възможността за лечение.
Промени в костите и меките тъкани
В костната тъкан се откриват промени, подобни на проявите на хипопаратироидизъм:
- дифузна остеопороза;
- кисти (кафяви или гигантоклетъчни тумори).
Калцият се освобождава от костите и образува калцификации в меките тъкани:
- в подкожната тъкан;
- мускули, включително миокарда;
- бъбреци;
- в роговицата и конюнктивата на очите;
- стените на главните артерии.
По кожата се появяват области на хиперпигментация.
Други знаци
Нарушаването на калциевия метаболизъм води до гърчове и повръщане. В някои случаи се наблюдава хематурия поради образуването на оксалатни кристали в пикочните пътища.

Умствена изостаналост, диабет, катарактата също може да бъде проява на болестта на Олбрайт.
Диагностика
Диагнозата се основава на анализ на клиничните прояви, рентгеново изследване на костите и меките тъкани и откриване на псевдохипопаратироидизъм с помощта на биохимични тестове.
Кръвните изследвания показват хипокалциемия и хиперфосфатемия, нивата на серумната алкална фосфатаза са нормални или повишени, както и PTH.
Лабораторно доказателство за псевдохипопаратиреоидизъм е липсата на повишаване на нивото на нефрогенния сАМР и фосфат в урината в отговор на прилагането на паратиреоиден хормон.
Възможности за лечение
За разлика от истинския хипопаратиреоидизъм, при болестта на Олбрайт приложението на паратироиден хормон няма ефект.
Целта на лечението е да се възстановят нормалните нива на калций и витамин D в кръвта. За да направите това, предписвайте лекарства, съдържащи тези вещества: дихидротахистерол, оксидевит, калцитрин.
Catad_tema Педиатрия - статии
Псевдохипопаратироидизъм (наследствена остеодистрофия на Олбрайт): трудности при диференциално диагностично търсене. Клинично наблюдение
Е.В. Тозлиян, детски ендокринолог, генетик, д-р И.В. Шулякова, невролог, д-р,
изолиран структурно подразделение"Научно-изследователски клиничен институт по педиатрия" Държавна бюджетна образователна институция за висше професионално образование "Руски национален изследователски медицински университет на името на N.I. Пирогов" на Министерството на здравеопазването на Руската федерация, Москва
Ключови думи:
деца, псевдохипопаратиреоидизъм, наследствена остеодистрофия на Олбрайт, затлъстяване, хипокалциемия, диагноза, резистентност към паратхормон.
Ключови думи:деца, псевдохипопаратиреоидизъм, наследствена остеодистрофия на Олбрайт, затлъстяване, хипокалцемия, диагностика, резистентност към паратиреоиден хормон.
Псевдохипопаратиреоидизъм (на гръцки pseudes - фалшив + хипопаратиреоидизъм; синоним: наследствена остеодистрофия на Олбрайт, синдром на яванското пиле) е рядко наследствено заболяване на скелетната система, симулиращо хипопаратиреоидизъм и характеризиращо се с нарушен метаболизъм на калций и фосфор; често придружени от забавено умствено и физическо развитие. Заболяването е описано за първи път от американския ендокринолог Олбрайт Ф. през 1942 г. Разпространението на заболяването е 7,9 на 1 милион души.
ГЕНЕТИЧНИ ДАННИ
Псевдохипопаратироидизмът (PHP) е генетично хетерогенно заболяване. Данните за вида на наследственото предаване са противоречиви: както Х-свързан доминантен, така и автозомно-доминантен, автозомно-рецесивен тип. В повечето случаи развитието на наследствената остеодистрофия на Олбрайт се свързва с мутации в локуса 20q13 на гена GNAS1, разположен на хромозома 20 (Patten et al., 1990), кодиращ Gs-алфа протеина, свързан с рецептора на паратироидния хормон (PTH). . Подобен фенотип беше открит и при пациенти с интерстициална делеция на дългото рамо на хромозома 2 в локуса 2q37.
ПАТОГЕНЕЗА
Патогенезата на псевдохипопаратиреоидизма се основава на генетично обусловена резистентност на бъбреците и скелета към действието на паратиреоидния хормон в резултат на дефект в комплекса „специфичен циторецептор – паратиреоиден хормон – аденилатциклаза“, който нарушава образуването на цикличен 3"- , 5"-аденозин монофосфат (сАМР) в бъбреците, който е вътреклетъчен медиатор на действието на паратироидния хормон върху метаболитните процеси. Псевдохипопаратиреоидизмът е генетичен хетерогенно заболяване.При някои пациенти самият циторецептор, който свързва паратироидния хормон, е дефектен (псевдохипопаратиреоидизъм тип 1А); други имат дефект в нуклеотид-свързващия протеин, локализиран в липидния двоен слой клетъчната мембранаи функционално свързване на рецептора с аденилат циклаза (псевдохипопаратироидизъм тип 1В). Някои пациенти имат ензимен дефицит на самата аденилат циклаза (тип 2 псевдохипопаратироидизъм). Дефицитът на cAMP в резултат на тези дефекти води до нарушаване на синтеза на специфични протеини, които определят биологичния ефект на паратироидния хормон. По този начин се губи чувствителността на целевите органи към паратироидния хормон.
КЛИНИЧНА ХАРАКТЕРИСТИКА
В момента има 4 клинични форми на патология: типове 1A, 1B, 1C и 2. Познаване на техните клинични и биохимични характеристики и данни генетични изследваниядава възможност за диференциална диагноза в рамките на самата нозологична форма.
Честите признаци, които позволяват да се подозира заболяването, са диспропорционалност във физическото развитие, нисък ръст (до степен на нанизъм) поради скъсяване на долните крайници (снимка 1), брахидактилия (снимка 2) и кръгла „лунообразна форма“ лице (снимка 3). Понякога се наблюдават екзостози и зъбна аплазия.
Снимка 1.
Външен виддете с остеодистрофия на Олбрайт
(характеристики на фенотипа, нисък ръст поради скъсяване на долните крайници)

Снимка 2.
Характеристики на скелетната система на пациента
с остеодистрофия на Олбрайт
(брахидактилия - скъсяване на пръстите)
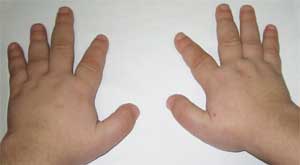
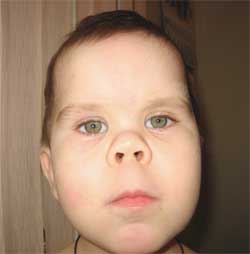
Снимка 3.
Характеристики на фенотипа на детето
с остеодистрофия на Олбрайт
(кръгло лице с форма на луна)
Патогномоничен признак е рязкото скъсяване на I, III и V метакарпалите и метатарзални кости(особено III и IV), в резултат на което вторите пръсти на ръцете и краката са по-дълги от останалите, а когато ръката е свита в юмрук, няма издутини в областта на IV и V метакарпофалангеални стави - т. нар. брахиметафалангизъм. Откриват се също къси широки фаланги, удебеляване на черепния свод и деминерализация на костите (остеопороза), затлъстяване.
Умствена изостаналост (обикновено умерена тежест) се установява при приблизително 20% от пациентите. Според някои автори умствената изостаналост се среща в 70% от случаите при наличие на хипокалциемия и в 30% от случаите при нормокалцемия. Психични процесипри пациентите се забавят. В неврологичния статус често се отбелязват двигателна неловкост и невротични реакции: страхове, тревожност, безпокойство, лош сън, повишени рефлекси, конвулсии с тетаничен характер и причинени от хипокалцемия, понякога конвулсивни пароксизми. Описани са и миопатични симптоми: мускулна умора, мускулна слабост. Често се наблюдават екстрапирамидни нарушения: хореиформна хиперкинеза, атетоза, лицев хемиспазъм, паркинсонизъм; в някои случаи се появяват епилептични пароксизми, церебеларни симптоми: атаксия, загуба на координация.
Често се откриват калцификации на меките тъкани и подкожни калцификации (гърди, корем, сухожилия на петата), като хистологичното им изследване показва остеома кутис(Izraeli et al., 1992), мозък (базални ганглии). Важно е да се отбележи, че калцификатите може вече да са налице при раждането. В резултат на хипокалцемия обикновено се развива катаракта и възникват дефекти на зъбния емайл.
ПСЕВДОХИПОПАРАТРОЗА ТИП 1А
има автозомно доминантен модел на наследяване. Генът за псевдохипопаратиреоидизъм тип 1А - GNAS1 - е локализиран на дългото рамо на хромозома 20, в локуса 20q13.2. Развитието на заболяването е свързано с дефицит на гуанин нуклеотид-свързващ протеин (Gs протеин). В същото време PTH, свързвайки се с рецепторите на целевите тъкани, не е в състояние да активира цикличния аденозин монофосфат (cAMP) и да предизвика тъканен отговор. Вероятно подобен механизъм е в основата на развитието на нечувствителност на тъканите на други органи и ендокринни жлези (хипофункция на щитовидната жлеза, половите жлези, хипофизната жлеза, захарен диабет, както и намалена реакция на черния дроб към приложението на глюкагон), наблюдавана при псевдохипопаратироидизъм тип 1А. При този тип патология не се наблюдава нормалната повишена екскреция на сАМР в урината в отговор на екзогенно приложение на ПТХ. Заболяването се диагностицира по-често на възраст 5-10 години. Болните са с нисък ръст, къс врат, кръгло лице, скъсяване на метакарпалните и метатарзалните кости (обикновено скъсяване на четвъртия и по-рядко втория пръст) - така нареченият брахи-метафалангизъм. Отбелязва се калцификация на меките тъкани и подкожни калцификации, които могат да бъдат открити при раждането; често се наблюдава едновременно засягане на други ендокринни жлези: щитовидна жлеза (хипофункция), гонади, панкреас (захарен диабет). В резултат на хипокалцемия често се развиват катаракта и дефекти на зъбния емайл. Като диференциално диагностичен тест за разграничаване на PHP тип 1А от хипопаратироидизъм: липса на клиничен ефект от парентерално приложениеПТХ под формата на повишаване на нивата на калций в кръвта и увеличаване на бъбречната екскреция на фосфор в урината (фосфатурен ефект).
При биохимични изследванияОткриват се хипокалцемия, хиперфосфатемия, повишени нива на паратироидния хормон в кръвта и хипофосфатурия. Нивото на Gs протеин в кръвта е намалено. При рентгеново изследванеСкелетната система разкрива скъсяване на метакарпалните и метатарзалните кости, генерализирана деминерализация и удебеляване на костите на черепния свод.
ПСЕВДОХИПОПАРАТРОЗА ТИП 1Б
има автозомно доминантен тип наследяване, но не е изключен доминантен, Х-свързан тип наследяване. Необходимо е да се има предвид наблюдаваното понякога непълно проникване на гена на заболяването и възможността за латентно носителство на патологията. Поради това се препоръчва клинично (откриване на субклинично протичане на заболяването) и биохимично изследване (определяне на нивата на калций, фосфор, ПТХ в кръвта) на предполагаеми носители на заболяването. PHP тип 1B се причинява от дефицит на тъканни рецептори за паратиреоиден хормон в прицелните органи и ограничена резистентност към паратиреоиден хормон. Клиничната картина е подобна на тази при тип 1А, но няма увреждане на други ендокринни жлези, по-рядко се среща остеодистрофия.
Пациентите нямат бъбречен отговор на екзогенно приложение на паратиреоиден хормон под формата на повишена екскреция на цикличен аденозин монофосфат в урината; обаче, за разлика от тип 1А, нивото на Gs протеин в кръвта е нормално. Жените са засегнати по-често от мъжете, но тежестта на заболяването може да бъде еднаква както при мъжете, така и при жените.
ПСЕВДОХИПОПАРАТРОЗА ТИП 1C
някои автори го идентифицират с псевдо-псевдохипопаратироидизъм (PPHP), описан от Albright F. през 1952 г. Характеризира се с клинична картина, характерна за PHP, но нивата на калций и фосфор в кръвта и урината остават в нормални граници. Нивата на PTH и Gs протеин в кръвта също остават на нормални нива. Някои пациенти с PHP тип 1C имат делеции de novoна хромозома 2. Възможно е този вариант на заболяването да е подвид на PGP тип 1А.
ПСЕВДОХИПОПАРАТРОЗА ТИП 2
клинично подобен на други видове заболяване, но има автозомно-рецесивен начин на наследяване. Не може да се изключи наличието на автозомно-доминантни форми на патология. Патогенезата на развитието е свързана с вътреклетъчна резистентност към сАМР. След това PTH се свързва с рецепторите и предизвиква нормален клетъчен отговор към PTH под формата на повишена екскреция на cAMP. Вътреклетъчната нечувствителност към сАМР обаче не позволява да се реализира пълният ефект на ПТХ. В същото време нормалната реакция на бъбреците към екзогенно приложение на паратиреоиден хормон се поддържа под формата на повишена екскреция на цикличен аденозин монофосфат в урината. Предполага се, че PHP тип 2 може да бъде свързан с дефицит на витамин D.
По този начин идентифицираните видове PGP се характеризират клинично с намалена чувствителност на целевите органи към ПТХ, но се различават по патогенетичните механизми на образуване на тъканна нечувствителност.
ДИАГНОСТИКА
Лабораторен диференциално диагностичен тест може да бъде моделът на бъбречна екскреция на сАМР в отговор на прилагане на ПТХ: повишена екскреция на сАМР се наблюдава при тип 2 и липсата му при тип 1. Диагнозата се потвърждава от откриването на намалено ниво на гуанин нуклеотид свързващ протеин (Gs протеин) в кръвта (средно 1,5-2 пъти) в сравнение с нормата. Хипокалциемията обикновено се комбинира с хиперфосфатемия и хипофосфатурия. Нивата на ПТХ са повишени; при тип 1C нивото на ПТХ е нормално, което води до името „псевдохипопаратироидизъм“. Рентгеновото изследване на скелетната система разкрива скъсяване на метакарпалните и метатарзалните кости, често генерализирана деминерализация (остеопороза) и удебеляване на костите на черепния свод. Дерматоглифният модел показва изместване на аксиалния палмарен трирадиус.
Критерии за диагностика:
- нисък ръст;
- кръгло лице;
- забавено нервно-психическо развитие;
- скелетни аномалии;
- нисък серумен калций;
- високо ниво на паратхормон в кръвта;
- намалена уринна екскреция на фосфати и сАМР.
ЛЕЧЕНИЕ И ПРОФИЛАКТИКА
Лечението на хипокалцемия се състои в предписване на калциеви добавки в дози, достатъчни за поддържане на нормални концентрации на калций в кръвта. Голямо значениеима терапия с витамин D. Понастоящем се използват активни метаболити на витамин D - оксидевит, 1-алфа-D3, калцитрин и др., в доза 1-2 mcg/ден с положителен резултат(повишени нива на калций в кръвта, намалени прояви на конвулсивен синдром). Тахистин (0,5–1,5 mg/ден) също е ефективен. Това лекарство повишава абсорбцията на калций в червата и по този начин спомага за повишаване на нивото на калций в кръвта. Антиконвулсивната терапия се използва като допълнителна терапия. На интелектуално развитиелечението няма забележим ефект, но заедно с намаляването на симптомите на конвулсивния синдром се наблюдава регресия на неврологичните прояви (подкорови нарушения, хореиформна хиперкинеза, атетоза и др.). За да се избегне предозиране на препарати с витамин D, е необходимо да се следи концентрацията на калций в кръвта на всеки 3-7 дни през първите 2 седмици от лечението и всеки месец през следващите 2-3 месеца. След достигане на стабилна концентрация на калций в кръвта е достатъчно да се проверява веднъж на всеки 2-3 месеца. Диета с ограничен прием на фосфор помага за нормализиране на нивата на фосфор и калций в кръвта и облекчава симптомите вторичен хиперпаратироидизъм. В случай на недостатъчност на други ендокринни жлези, заместителна терапиясъответните хормони.
Лечението с паратироиден хормон е неефективно. За облекчаване на конвулсивни атаки се прилага интравенозно 10% разтвор на калциев хлорид или калциев глюконат; перорално - 5-10% разтвор на калциев хлорид, 1 супена лъжица 3-4 пъти на ден: калциев глюконат, калциев лактат - до 10 g на ден.
ПРОГНОЗАза цял живот се определя от тежестта на конвулсивния синдром.
ПРЕДОТВРАТЯВАНЕзаболяване се основава на данни от медицинско генетично консултиране.
МЕДИКО-ГЕНЕТИЧНО КОНСУЛТИРАНЕ
При провеждане на медицинско генетично консултиране трябва да се изхожда от автозомно-доминантния тип наследство и високия (50%) риск от повторение на заболяването в семейството с наследствени форми. За да се определи естеството на вида на наследството, е необходимо да се извърши задълбочен преглед на родителите, тъй като синдромът може да се прояви минимално клинични симптоми. Понастоящем е разработена и усъвършенствана молекулярно-генетична диагностика на заболяването чрез типизиране на мутации в гена GNAS1 на хромозома 20. Разработват се методи за пренатална диагностика на заболяването като цяло и неговите отделни видове.
КЛИНИЧНО НАБЛЮДЕНИЕМомче Г., 14,5 години (снимка 4), е прието в Научноизследователския клиничен институт по педиатрия с диагноза дегенеративно заболяване нервна система? вродена външна хидроцефалия; симптоматична епилепсия; наследствен синдром? болест на съхранението? метаболитна енцефалопатия; субклиничен хипотиреоидизъм; нисък ръст смесен произход; когнитивно увреждане.
Оплакванияпри приемане на интензивни пароксизмални главоболия, локализирани в челната област и придружени от повръщане, което носи облекчение, намалена памет и ефективност в училище, конвулсивни атаки, по време на които се появяват потрепвания в дясната ръка.
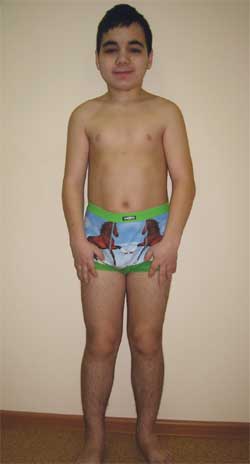
Снимка 4.
Дете Г., 14,5 години, с остеодистрофия на Олбрайт
(фенотипни характеристики, нисък ръст, скъсени крайници, брахидактилия)
Семейна история:родителите са арменци по националност, нямат кръвна връзка и нямат професионални рискове. В родословието няма случаи на психични заболявания, епилепсия или изоставане в развитието. Sibs, сестра, на 17 години, се казва, че е здрава.
История на живота и болестта:момче от 2-ра бременност, протекла без особености, второ раждане, на термин, физиологично, тегло - 3100 гр., дължина - 51 см. Извика веднага, оценка по Апгар - 7/9 точки. Влошаване на състоянието на 3-ия ден - неонатални гърчове, спрени в родилния дом. Ранният постнатален период е без особености. Имаше леко забавяне в двигателното развитие през първата година от живота, независимо ходене от 1 година 3 месеца. Във връзка с това е наблюдаван от невролог с диагноза органично увреждане на централната нервна система; вродена хидроцефалия; неонатални гърчове; анамнеза за фебрилни гърчове.
Получих диакарб и финлепсин. Начало на атаките на 1 година и 11 месеца. – асиметричен, тонизиращ под формата на напрежение дясна ръкаи краката, с отваряне на очите, до 2 минути, без загуба на съзнание, често до 10 епизода на ден. Получавах Депакин нередовно. На фона на независимо оттегляне - единичен тонизиращ статус. На 2 години е направена компютърна томография на мозъка по местоживеене, където изолирани огнищадемиелинизация в тилни дялове.
Направена е консултация с неврохирург и е препоръчано консервативно лечение. От 3-годишна възраст се наблюдава изоставане в психо-речевото развитие и се препоръчва наблюдение от психиатър.
От 4-5-годишна възраст родителите започват да забелязват деформация и скъсяване на пръстите на ръцете и краката, особено на 2-4 пръста симетрично на ръцете и краката, както и намаляване на показателите за растеж. На 8-годишна възраст заключението на логопеда е общо нарушение на речта от 2-3-то ниво, препоръчва се обучение в специализирано училище. На същата възраст преглед от генетик по местоживеене, заключение: наследствено заболяванеобмен? Препоръчва се изследване на кръвните аминокиселини, не са открити промени; крайно заключение: не са установени данни за наследствено метаболитно заболяване; хипохондроплазия; Препоръчва се лечение при невролог и ендокринолог.
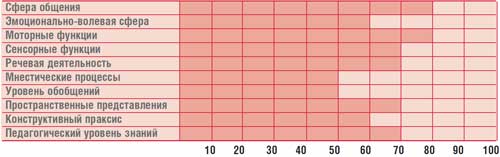
Таблица.
Профил на умственото развитие на дете Г., 14,5 години (IQ = 68)
На 8 години е консултиран от ендокринолог за забавен растеж и развитие. При рентгеново изследване на ръцете се установяват следните особености: средни, основни фаланги и метакарпални костискъсен, удебелен; Диагнозата на рентгенолога е ахондроплазия.
Многократно прегледан по местоживеене в неврологична болница. На 12-годишна възраст се появиха конвулсивни пристъпи без загуба на съзнание с потрепвания на дясната ръка, които бяха серийни, беше назначена антиконвулсивна терапия (депакин), честотата на пристъпите значително намаля. На 13 години е направен ЯМР на мозъка с контраст - симетрични изменения в основата темпорални дяловена ядрено ниво под формата на повишаване на MR сигнала, което е типично за токсични (манган) или метаболитни (мед, желязо) енцефалопатии.
Прегледана на 13 години 3 месеца. ендокринолог, изследването на профила на щитовидната жлеза разкрива увеличение тироид-стимулиращ хормон(TSH), диагностициран е субклиничен хипотиреоидизъм, предписан е L-тироксин.
При анализа на амбулаторната карта и документацията на детето по местоживеене е проведено изследване на калций и фосфор веднъж, на възраст от 1,5 години, е отбелязана хипокалциемия, но не е извършено допълнително изследване по този повод. Като се има предвид несигурността на диагнозата по местоживеене, генетикът изпрати детето в Москва, в Научноизследователския клиничен институт по педиатрия, за да изясни диагнозата.
Данни от обективно изследване:
Височина – 143 см, тегло – 43 кг.
Физическо развитиемного ниско, хармонично, непропорционално телосложение поради скъсени крайници. Растежът Sds съответства на –2,8 отклонения от нормата (норма –2+2).
Фенотипни характеристики: кръгло лице, къса шия, антимонголоидни палпебрални фисури, широк мост на носа, високо чело, брахидактилия, скъсяване на IV и V метакарпални и метатарзални кости (снимка 5). Вътрешни органи – без особености. Сексуално развитие – етап III–IV по Танер (което съответства на възрастта).
Данни от лабораторни и функционални изследвания:
Клиничен анализкръвта и урината са нормални.
Биохимичен кръвен тест: общ калций – 1,39 (норма 2,02–2,6 mmol/l), йонизиран калций – 0,61 (норма 1,13–1,32 mmol/l), неорганичен фосфор – 3,66 (норма 0,86–1,56 mmol/l), останалите показатели са в рамките нормални граници.
Биохимичен анализ на урината: бъбречната екскреция на фосфати е намалена - 11,5 mmol/l (норма 19–32 mmol/l).
Профил на щитовидната жлеза: TSH – 11,75 (нормален диапазон 0,4–4,0 µIU/ml), свободен Т4 – 0,49 (нормален диапазон 1,0–1,8 ng/dL).
Паратироиден хормон – 499 (норма 12–65 pg/ml), STH – 7 ng/ml (норма 7–10 ng/ml), соматомедин-С – 250 ng/ml (норма 88–360 ng/ml).
Ехография на вътрешни органи - без особености.
ЕКГ - миграция на суправентрикуларния пейсмейкър на фона на нормална сърдечна честота от 71-80 удара / мин. Непълна блокада десен кракНеговият пакет. Нарушаване на процеса на реполяризация в миокарда на задната стена на лявата камера (намаляване на ZT III, aVF).
R-графия на гръбначния стълб – дясностранна сколиоза гръднигръбначен стълб 1-ва степен, тежка остеопороза.
R-графия на ръцете с улавяне на предмишниците - скъсяване и разширяване на крайните и средните фаланги. Костна възраст – 13,5–14 години.
Не са регистрирани ЕЕГ модели на епилептична активност.
ЯМР на мозъка - ЯМР картина на множество подкорови огнища на повишен МР сигнал в фронтални дялове, външна компенсирана хидроцефалия с атрофия на мозъчното вещество.
MSCT на мозъка показва симетрични области на калцификация на лентиформени ядра. Дифузни хиперденсни зони в таламуса, опашните ядра с област на калцификация вдясно. Множество точкови калцификации на покривните меки тъкани на черепа.
Аудиограма – без патология.
В ход е ДНК диагностика в гена GNAS1.
Специализирани консултации:
Ендокринолог – Наследствена остеодистрофия на Олбрайт тип 1А (псевдохипопаратироидизъм). Първичен хипотиреоидизъм, непълна лекарствена компенсация.
Офталмолог – пълна вторична катаракта. Препоръчва се хирургично лечение.
Психолог – когнитивно увреждане (психологичният профил на детето е представен в таблицата).
Като се вземат предвид фенотипа на детето, медицинската история, резултатите от допълнителни изследвания (хипокалциемия, хиперфосфатемия, хипофосфатурия, повишен паратиреоиден хормон в кръвта), калцификации в мозъка, наличие на катаракта, хипотиреоидизъм), се поставя диагноза: наследствена остеодистрофия на Олбрайт. тип 1А (псевдохипопаратироидизъм). Препоръчва се провеждането на ДНК диагностика - търсене на мутации в гена GNAS1.
Лечение:Детето се препоръчва да приема еутирокс в доза 100 mcg/ден; активен метаболит на витамин D - алфа-D3 (Teva) в доза 2 мкг/ден; калций (Sandoz) 2000 mg/ден; постоянно прилагане на антиконвулсивна терапия - финлепсин 800 mg/ден под наблюдението на невролог-епилептолог; занимания с логопед и психолог; енерготропна терапия (Elkar и коензим Q10 в дози, свързани с възрастта). Проследяване на показателите на фосфорно-калциевия метаболизъм, нивата на паратироидния хормон.
По този начин,Представеното клинично наблюдение показва трудностите на диференциално-диагностичното търсене, важността на навременното изследване на прости биохимични параметри (в случай на епилепсия е задължителен повторен скрининг на показателите на фосфорно-калциевия метаболизъм), резултатите от късната диагностика на генетично обусловено заболяване , необходимостта от интегриране на отделни признаци в общия фенотип на определено заболяване патологично състояниеза насочена, навременна диагностика отделни форми наследствени заболявания. Навременната диагностика и изясняване на генезиса на всеки синдром са особено важни, тъй като ни позволяват да намерим оптималния подход за лечение на тези състояния и профилактика. възможни усложнения(до увреждане на детето); предотвратяване на повторна поява на наследствени заболявания в засегнатите семейства (медико-генетично консултиране). Това налага необходимостта лекарите от различни специалности ясно да се ориентират в потока на наследствено обусловената патология. Списъкът с литература е в редакцията.

