HEREDITARY DISEASES NG NERVOUS SYSTEM
LECTURE 16
Ang mga degenerative na sakit na may isang nangingibabaw na sugat ng neuromuscular apparatus ay bumubuo sa pinakamalaking grupo ng lahat ng mga namamana na sakit.
Lubhang mahalaga, at madalas na mapagpasyahan sa pagsusuri ng mga sakit na neuromuscular, ay ang mga resulta ng electrophysiological at biochemical na pananaliksik. Ang kahalagahan ng mga natuklasang pathomorphological ay kasinghusay din. Ang pag-aaral ng biopsy ng kalamnan sa ilalim ng isang ilaw na mikroskopyo ay nakakatulong na makilala ang myogenic mula sa neurogenic atrophy. Ang pagsusuri sa histochemical ay kinakailangan upang makita ang mga metabolic na sugat sa kalamnan, at mikroskopya ng elektron natuklasan ang isang buong malaking klase ng mga sakit - non-progressive myopathies.
Ang banayad na pag-uunat ng mga apektadong kasukasuan ay binabawasan ang paglitaw ng mga contracture at sa gayon ay humahantong sa isang mas mabagal na pag-unlad ng sakit. Sa kasong ito, ang iba't ibang mga partikular na ehersisyo ay isinasagawa, at ang Urias pressure bandage ay napatunayang epektibo sa kaso ng kapansanan sa lalim ng paa at madalas na itinuturing na paggamot sa bahay sa paglabas. Ang pagsasanay sa sensorimotor ng mga pasyente na may mga sakit sa kalamnan ay isinasagawa sa indibidwal o grupo na therapy at nakatuon sa koordinasyon at mahusay na mga kasanayan sa motor.
napaka mahalagang pamamaraan paggamot para sa mga taong may sakit sa kalamnan ay upang maibalik ang kakayahang magmaneho muli ng kotse. Para sa layuning ito, mayroong pakikipagtulungan sa isang panlabas na paaralan sa pagmamaneho sa departamento ng occupational therapy at ang pagmamaneho sa Bad Wildungen ay isinasagawa sa isang angkop na sasakyang may kapansanan na may sinanay na tagapagturo sa pagmamaneho.
Mga progresibong muscular dystrophies. Ang terminong muscular dystrophies ay isang pangkat ng mga genetically determined disorder na nailalarawan sa pamamagitan ng progresibo degenerative na pagbabago sa mga fibers ng kalamnan na walang pangunahing patolohiya ng peripheral (mas mababang) motor neuron.
Ang iba't ibang mga anyo ay naiiba sa bawat isa sa mga uri ng mana, ang tiyempo ng pagsisimula ng proseso, ang kalikasan at bilis ng kurso nito, ang kakaiba ng topograpiya ng pagkasayang ng kalamnan, ang pagkakaroon o kawalan ng pseudohypertrophy at pagbawi ng tendon, at iba pa. palatandaan.
Ang isang karagdagang diskarte sa rehabilitasyon ng mga sakit sa kalamnan ay ang pagkakaloob ng sapat na pagpapayo. Nalalapat ito hindi lamang sa mga orthoses, ngunit sa lahat ng iba pa AIDS sa bahay, Ang naka-target na tulong sa mga taong may kapansanan na may mga sakit sa kalamnan ay lubhang kapaki-pakinabang, ang mga wheelchair ay tumpak na sinusukat at nababagay sa mga pangangailangan ng mga taong dumaranas ng mga sakit sa kalamnan. Shower chair, bath lift, wheelchair, rampa para sa mga wheelchair at iba pa.
Gumagamit ang Kagawaran ng Physical Therapy ng iba't ibang pamamaraan para ma-relax ang pasyente. Bilang karagdagan, ang departamento ng paggamot na ito ay maaaring mabawasan ang madalas na maliliit na sintomas ng sakit sa mga pasyente na may mga sakit sa kalamnan. Ang speech therapy ay isa pang departamento ng rehabilitasyon para sa paggamot ng mga sakit sa kalamnan. Ang isa pang diagnosis ay dapat ding gawin dito gamit ang laryngoscopy o tiyak pag-aaral ng x-ray. Sa isang kwalipikadong klinika sa rehabilitasyon para sa paggamot ng mga sakit sa kalamnan, ang isang holistic na therapeutic approach sa sakit na ito ay isang mahalagang kinakailangan.
Karamihan sa mga muscular dystrophies ay mahusay na pinag-aralan sa klinikal, sila Detalyadong Paglalarawan ginawa sa katapusan ng huling siglo. Ngunit, sa kabila ng halos isang siglo ng kasaysayan ng pag-aaral ng myodystrophy, ang mga isyu ng kanilang pathogenesis at paggamot ay nananatiling hindi nalutas hanggang sa araw na ito. Malaki ang pag-asa sa molecular genetics, sa tulong kung saan ang lokasyon ng mga gene ng maraming nosological form ay natukoy na.
Kabilang dito ang nagtapos na mga mag-aaral sa sikolohiya ng faculty. Ang mga apektadong tao ay nagdurusa dahil sa pisikal na kakulangan at kaugnay na mga kahihinatnan sa pribado - Ang pag-asa sa buhay ay kadalasang nauugnay sa depressive moods at mga reklamong psychosomatic. Ang pagsuporta sa mga indibidwal na pag-uusap o pag-aaral tungkol sa mga diskarte sa pagpapahinga gaya ng autogenic na pagsasanay o progresibong pagpapahinga ng kalamnan ay lubhang nakakatulong dito.
Mga tanong tungkol sa batas sa pag-aalaga, malubhang kapansanan, lugar ng trabaho, pakikipag-ugnayan sa isang tagapag-empleyo para sa bokasyonal na rehabilitasyon o pagsasama sa isang kumpanya ay dapat na banggitin dito bilang madalas na mga paksa sa pagpapayo sa lipunan. Bilang karagdagan, ang naka-target na pagpapayo sa mga konsultasyon ay madalas madalas itanong para sa mga pasyente na may mga sakit sa kalamnan. Sa wakas, kasama rin sa plano ng therapy sa panahon ng rehabilitasyon sa Bad Wildungen ang koordinasyon ng isang kwalipikadong espesyalista sa orthopaedic. Si Vicerek ay ang punong manggagamot ng orthopaedic department ng klinika sa Homberg sa Bad Wildungen.
Ang diagnosis ng muscular dystrophies ay kadalasang nagdudulot ng malaking kahirapan. Mayroong malaking pagkakaiba-iba sa mga klinikal na pagpapakita, at ang isang maliit na bilang ng mga miyembro ng pamilya ay nagpapahirap sa pagtukoy ng uri ng mana.
Ang isang katangian na depekto sa motor sa mga pasyente na may muscular dystrophies ay isang "duck" na lakad: ang pasyente ay naglalakad na paikot-ikot sa isang tabi. Ito ay pangunahing nauugnay sa kahinaan ng mga kalamnan ng gluteal, lalo na sa gitna at maliliit, na nag-aayos ng pelvis na may kaugnayan sa femur. Bilang resulta, ang sakit ay nagdudulot ng pagkiling ng pelvis patungo sa hindi nakasuportang binti (Trendelenburg phenomenon) at isang compensatory tilt ng katawan sa kabaligtaran(Duchenne phenomenon). Kapag naglalakad, ang gilid ng slope ay patuloy na nagbabago. Ang mga pagbabagong ito ay maaari ding suriin sa pagsusulit ng Trendelenburg sa pamamagitan ng pagtatanong sa pasyente na itaas ang isang paa, baluktot ito sa tamang anggulo sa mga kasukasuan ng tuhod at balakang: ang pelvis sa gilid ng nakataas na binti ay bumababa (at hindi tumataas gaya ng normal) dahil sa kahinaan ng gluteus medius na kalamnan ng sumusuporta sa binti.
Ang klinika ay nagsasagawa ng mga regular na pagsasanay, pati na rin ang pakikilahok sa mga kaugnay na kongreso at symposium. Ang pagpupulong na ito ay naganap, sa partikular. Kadalasan mayroon ding pagtaas sa mga enzyme ng kalamnan. Ang mga sakit ay kadalasang umuunlad nang dahan-dahang gumagapang, ngunit nangyayari ang mabilis na mga sakit. Kung pinaghihinalaan mo na mayroon kang myopathy o kasangkot sa isang kilalang sakit na neuromuscular, maaari kang gumawa ng mga appointment.
Ang paggamot ay nakasalalay sa tiyak na dahilan, nagpapaalab na sakit sa kalamnan ay maaaring, halimbawa, Halimbawa, iba't ibang mga immunotherapeutic agent. Detalyadong pangkalahatang-ideya ang iba't ibang genetic na sakit sa kalamnan ay matatagpuan sa mga talaan ng gene ng neuromuscular disease.
Bumangon mula sa isang pahalang na posisyon, ang isang pasyente na may malubhang kahinaan ng kalamnan ng mga proximal na kalamnan ay halos hindi gumulong sa kanyang tiyan, pagkatapos, ipinatong ang kanyang mga kamay sa sahig, nakadapa at pagkatapos, ipinatong ang kanyang mga kamay sa mga shins, pagkatapos ay sa mga balakang. , unti-unting umayos. Ang hindi pangkaraniwang bagay na ito ay tinatawag na Gowers maneuver. Kadalasan ito ay nauugnay sa kahinaan ng mga kalamnan ng gluteus maximus.
Mga gamot na maaaring magdulot o magpapataas ng sakit sa kalamnan
Ang lahat ng kinakailangang diagnostic na posibilidad ay magagamit sa aming pagsasanay. Ang pagsasanay ay kasangkot dito, ang regular na pagsasanay sa larangan ng mga sakit sa neuromuscular ay nakaayos. Ang mga apektadong tao ay maaari ding magparehistro sa rehistro ng pasyente. Ang mga pagpapatala ng pasyente ay isang mahalagang unang hakbang tungo sa mas mahusay na pag-unawa sa pananaliksik sa pagpapaunlad ng sakit at therapy.
Pinsala ng kalamnan ng puso
Ang posibleng pagkakasangkot ng kalamnan ng puso sa myopathy at iba pang mga sakit na neuromuscular ay sinusuri dito.Duchenne myodystrophy. Ang pseudohypertrophic Duchenne muscular dystrophy ay nangyayari nang mas madalas kaysa sa lahat ng iba pang sakit ng muscular system (30 sa bawat 100,000 live birth). Ito ay nailalarawan sa pamamagitan ng maagang simula at malignant na kurso. Ang klasikong larawan ay ipinakita sa pamamagitan ng isang pagbabago sa lakad sa isang bata na may edad na 2-5 taon, sa edad na 8-10 taon ang mga bata ay nahihirapan nang lumakad, sa edad na 14-15 sila ay karaniwang ganap na hindi kumikilos. Ang mga bata ay may higit pa maagang edad ang mga unang sintomas ay ipinakita sa pamamagitan ng isang lag sa pag-unlad ng motor: nagsisimula silang maglakad mamaya, hindi sila maaaring tumakbo at tumalon. Ang mga pasyente ay namamatay sa ika-2 o ika-3 dekada ng buhay.
Ang ibig naming sabihin ay lahat ng sakit sa neuromuscular. Ayon sa klasipikasyon ni Walton, mayroong 800 na anyo. Ang sintomas na ito ay maaaring limitado sa ilang grupo ng kalamnan, o, depende sa sakit, ang buong kalamnan ay maaaring matagpuan. Pag-aaksaya ng kalamnan, pagbabawas masa ng kalamnan At kahinaan ng kalamnan ay ang mga pangunahing sintomas ng mga sakit na neuromuscular. Ang mga gamot ay tinutukoy bilang pagbabawas ng musculoskeletal muscle atrophy. Gayunpaman, hindi pa sila nagbibigay ng direktang diagnosis. Mayroong ilang mga kadahilanan na maaaring humantong sa isang bahagyang pagbawas sa mass ng kalamnan, na bahagyang nauugnay sa buong katawan.
Ang isa sa mga unang palatandaan ng sakit ay ang compaction ng mga kalamnan ng guya at isang unti-unting pagtaas sa kanilang dami dahil sa pseudohypertrophy. Ang pagkasayang ng mga kalamnan ng hita, ang pelvic girdle ay madalas na natatakpan ng mahusay na binuo subcutaneous fatty tissue. Unti-unti, ang proseso ay tumatagal ng pataas na direksyon at kumakalat sa kabila ng sinturon ng balikat, mga kalamnan sa likod, at pagkatapos ay sa mga proximal na bahagi ng mga braso.
Ang tunay na sanhi ng naturang muscular atrophy o kahinaan ng kalamnan ay maaaring nasa malayo, ganap na magkakaibang bahagi ng katawan. Ang mga sanhi ay maaaring nasa mga selula ng nerbiyos spinal cord, responsable para sa paggalaw, sa supply nerves, kapag ang nerve ay inilipat sa kalamnan, o sa mga kalamnan mismo.
Paggamot ng mga sakit sa neuromuscular
May mga promising na gamot para sa paggamot ng myositis, myasthenia gravis, at endocrine myopathy. Ang mga unang diskarte sa pagbagal ay posible sa lateral sclerosis amyotrophic. Sa kaso ng hereditary muscular dystrophies at spinal muscular atrophies, ang isang causative treatment ay hindi pa naitatag. Ang mga espesyal na inaasahan para sa hinaharap ay nilalayon therapy ng gene. Sa lahat ng mga sakit na neuromuscular, ang symptomatology ay pare-parehong suporta sa physiotherapeutic kasama ng mga orthopedic na hakbang, sa ilang mga kaso na may suporta sa paghinga at sa ilang mga kaso na may posibilidad ng paglipat ng puso.
SA yugto ng terminal Ang kahinaan ng kalamnan ay maaaring kumalat sa mga kalamnan ng mukha, pharynx, mga kalamnan sa paghinga.
Sa advanced na yugto ng sakit, mayroong mga sintomas ng katangian, Paano" lakad ng pato”; binibigyang diin ang lumbar lordosis, pterygoid scapulae, sintomas ng "maluwag na sinturon sa balikat". Ang maagang pag-urong ng kalamnan at pag-urong ng litid ay karaniwan, lalo na sa mga tendon ng Achilles. Ang mga reflexes ng tuhod ay nahuhulog nang maaga, at pagkatapos ay mga reflexes mula sa itaas na mga paa't kamay.
Kasama rin sa paggamot ang pagbabayad para sa mga pisikal na limitasyon na may naaangkop na paraan. Ang German Society for Muscular Disease ay nag-set up ng isang independiyenteng resource center. Mayroon ding posibilidad na subukan ang mga produkto sa bahay sa dalawang karaniwang apartment o gumugol lamang ng ilang araw sa mga apartment na walang harang.
Mga Muscular Disorder Ang mga Independent Muscular Disorder ay medyo bihira. Sa kabilang banda, madalas na nangyayari na ang mga kalamnan ay apektado ng iba pang pinagbabatayan na sakit, lalo na ang mga sakit sistema ng nerbiyos; Nakakahawang sakit tulad ng tipus at tuberculosis; Mga sakit na parasitiko, Trinity at porcine worm, pati na rin ang mga hormonal disorder.
Ang pseudohypertrophy ay maaaring umunlad hindi lamang sa gastrocnemius, kundi pati na rin sa mga kalamnan ng gluteal, deltoid, tiyan, at dila. Kadalasan ang kalamnan ng puso ay naghihirap mula sa uri ng cardiomyopathy. Mga kaguluhan sa ritmo ng aktibidad ng puso, pagpapalawak ng mga hangganan ng puso, pagkabingi ng mga tono, Mga pagbabago sa ECG. Ang talamak na pagpalya ng puso ay ang pinaka parehong dahilan pagkamatay sa Duchenne myodystrophy. Sa autopsy, matatagpuan ang fibrosis at fatty infiltration ng kalamnan ng puso.
Ang simpleng pagkawala ng kalamnan ay kadalasang nangyayari bilang resulta ng pag-iingat o pahinga, Ang pag-urong ng kalamnan ay maaaring sanhi ng mga adhesion o pinsala dahil sa mahinang sirkulasyon - Bilang resulta ng mga asosasyon ng inis. Ang sanhi ng pananakit ng kalamnan ay maaaring rayuma ng kalamnan, sobrang pagod, sipon, metabolic disorder o labis na pagsusumikap ng ilang grupo ng kalamnan dahil sa deformity ng skeletal.
Myoma - benign tumor tissue ng kalamnan. Polymyositis: Sakit sa kalamnan na may katulad na mga sintomas sa dermatomyositis ngunit wala pantal sa balat. Kung tungkol sa dahilan, ipinapalagay na ang paglabag ay tumutukoy sa mga auto-aggression, bilang sa pagdurusa na nagmumula bilang resulta ng pag-atake ng katawan sa sarili nitong mga istruktura.
Kadalasan mayroong isang paglabag sa motility ng gastrointestinal tract.
Ang pagbaba ng katalinuhan ay isang pangkaraniwang sintomas. Ang interes ay ang katotohanan na sa ilang mga pamilya ang oligophrenia ay malinaw na ipinahayag, sa iba ay medyo katamtaman. Ang pagbabago sa mas mataas na pag-andar ng kaisipan ay karaniwang hindi umuunlad at hindi nauugnay sa kalubhaan ng depekto ng kalamnan. Hindi ito maipaliwanag lamang ng pedagogical na kapabayaan ng mga may sakit na bata, na maagang tumalikod sa mga grupo ng mga bata, ay hindi dumalo. kindergarten at paaralan dahil sa kapansanan sa motor. Ang CT at MRI ay madalas na nagpapakita ng cerebral atrophy, na posibleng nauugnay sa kapansanan sa pag-unlad ng prenatal ng utak.
Mayroong higit sa 200 mga uri ng sakit sa kalamnan, ang ilan ay pinangalanan sa kanilang mga natuklasan at ang iba naman ay ayon sa karamdaman. Ang pinakakaraniwan ay tatlong subgroup. Progressive muscular dystrophy Spinal muscular atrophy Neuronal muscular atrophy. Sa klinikal na kasanayan, ang mga sakit na ito ay bihira at ganap na kinakailangan sa mga kamay ng isang neurologist.
Ang isang genetic defect sa chromosome 19 ay isang lesyon mga selula ng kalamnan na hindi magagamot hanggang ngayon. Ang dahilan ay malamang na isang pagbabago sa sistema ng lamad ng mga selula ng kalamnan. Ang mystic dystrophy ay maaaring umunlad sa kumpletong pagkasira ng mga selula ng kalamnan. Una, ang mga kalamnan ay apektado sa mukha, braso, bisig, ibabang binti at paa. Ang sakit ay maaaring mangyari sa mga kalalakihan at kababaihan sa lahat ng edad.
Kadalasan, ang mga bata ay nagkakaroon ng adiposogenital syndrome, minsan iba pang mga palatandaan ng kakulangan sa endocrine. Madalas makakita ng mga pagbabago sa skeletal system: deformity ng paa, dibdib, gulugod, nagkakalat ng osteoporosis.
Natatanging katangian Ang anyo ng Duchenne ay mataas na antas naka-on na ang hyperenzymemia maagang yugto pag-unlad ng proseso. Kaya, ang antas ng isang enzyme na tiyak para sa tissue ng kalamnan - creatinine phosphokinase - sa serum ng dugo ay maaaring lumampas sa sampu at kahit na daan-daang beses normal na pagganap. Ang isang matalim (10-100 beses) na pagtaas ng creatinine phosphokinase (CPK) sa neuromuscular pathology ay dapat mag-udyok ng talakayan lalo na sa mga sumusunod na sakit: Duchenne disease, Becker's disease, poliomyositis at dermatomyositis, paroxysmal myoglobulinuria, distal myodystrophy. Sa mga advanced na yugto lamang ng sakit, ang antas ng hyperenzymemia ay unti-unting bumababa. May mga ulat ng pagtaas ng CPK sa yugto ng pag-unlad ng intrauterine.
Ang pagkaantala ng pagpapahinga ng kalamnan ay karaniwang pagkatapos pag-igting ng kalamnan. Ang mga kahihinatnan ay kahinaan ng kalamnan at limitasyon ng paggalaw sa mga binti, braso at kamay, pagkasira ng mahusay na mga kasanayan sa motor. Ang ilang mga namamagang kalamnan ay lalong malakas dahil ang tipikal na komposisyon ng tissue ng pinagbabatayan mga hibla ng kalamnan naka-embed sa taba at connective tissue.
Ito ay tinutukoy din bilang ang spinal cord na nakakaapekto pamumulikat ng kalamnan. Mayroong hanggang 30 iba't ibang anyo spinal muscular atrophy. Ang pinakakaraniwang anyo ay proximal spinal muscular atrophy, na inilarawan dito. Ito ay itinalaga bilang mga sumusunod pagkatapos ng simula ng fuselage.
Ang Duchenne muscular dystrophy ay nakukuha sa isang X-linked recessive na paraan. Ang gene ay matatagpuan sa maikling braso ng X chromosome. Ang dalas ng mutation ng gene ay medyo mataas (30%), na nagpapaliwanag malaking bilang ng kalat-kalat na mga kaso.
Ang isang mutation (kadalasan ay isang pagtanggal) ay humahantong sa isang sekswal o halos kumpletong kawalan ng isang produkto ng gene - isang istrukturang protina ng dystrophic. Papel ng pisyolohikal Ang dystrophy ay hindi pa ganap na naitatag. Ito ay matatagpuan sa mataas na konsentrasyon sa sarcolemma, na tila gumaganap ng isang papel sa pagpapanatili ng integridad ng lamad na ito. Ang kawalan ng dystrophy ay sanhi mga pagbabago sa istruktura sa sarcolemma, na humahantong naman sa pagkawala ng mga bahagi ng intracellular at pagtaas ng pagpasok ng calcium, na sa huli ay humahantong sa pagkamatay ng myofibrils. Ito ay pinaniniwalaan na ang isang kakulangan ng dystrophic sa synaptic zone ng cortical neurons ay ang sanhi ng mental retardation.
Ang dahilan ay malamang na isang genetic defect. Mga selula ng nerbiyos sa spinal cord, ang mga anterior squamous cells ay nahawaan. Marahil, ang sanhi ay ang nervous system ng mga nerbiyos. Nalalapat lamang ito sa motor nervous system. Ang mga bahagi ng sistema ng nerbiyos na responsable para sa pandamdam ng pagpindot, pang-unawa sa sakit at temperatura ay nananatiling buo. Function Pantog at hindi lumalala ang tumbong.
Ang pagtatalaga ay kumakatawan din sa pagkawala ng mga kalamnan dahil sa mga ugat. Ang sanhi ay halos palaging isang genetic na depekto, ang mga kaluban ng mga nerve fiber ay nagiging abnormal na makapal, o ang mga nerve sheath mismo ay nawasak. Naka-on mga hibla ng nerve nakakaapekto sa mga kamay at paa.
Para sa medikal na genetic counseling, ang pagtatatag ng heterozygous carriage ay napakahalaga. Sa Duchenne myodystrophy sa heterozygotes, sa humigit-kumulang 70% ng mga kaso, subclinical, at kung minsan kahit na malinaw na mga palatandaan patolohiya ng kalamnan - ilang compaction at kahit na isang pagtaas sa mga kalamnan ng guya, mabilis na pagkapagod ng kalamnan sa panahon ng pisikal na pagsusumikap, mga pagbabago sa EMG at pathomorphological na pagsusuri ng mga specimen ng biopsy ng kalamnan. Kadalasan, ang mga heterozygous carrier ay nagpapakita ng pagtaas sa aktibidad ng creatinine phosphokinase.
Ang intensity ng bilis ng nerve conduction ay bumabagal. Nagsisimula sa mas mababang mga paa't kamay na may pagkawala ng kalamnan at kasabay na panghihina ng kalamnan. Ang mga sintomas pagkatapos ay tumaas sa mas mababang mga paa't kamay, sa kalaunan ay nakakaapekto sa mga braso at bisig. Mababa ang sensory disturbances. posibleng mga Autonomic na abala tulad ng labis o masyadong kaunting pagpapawis at pagkagambala sa pinagbabatayan ng daloy ng dugo ay posible. Maaaring mangyari ang mga menor de edad na spastic na sintomas sa mga binti.
Ang kahinaan ng kalamnan ay nagreresulta mula sa panghihina ng kalamnan hanggang sa ganap na gumana ang mga kalamnan, na nagreresulta sa makabuluhang matinding paghihigpit sa paggalaw sa mga binti, braso, at braso. Ang mga kalamnan ay isang napakahalagang bahagi ng ating katawan. Kung walang mga kalamnan, ang katawan ay nawawalan ng kakayahang kumilos at magsagawa ng iba't ibang mga aksyon. Sa katunayan, walang tao sistema ng mga kalamnan malamang hindi ka na makakaligtas. Ito ay dahil sa katotohanan na karamihan sa mga organo ay nasa sistema ng pagtunaw binubuo ng mga kalamnan at maging ang puso mismo, na nagbobomba ng dugo, ay isang kalamnan din.
Sa pagkakaroon ng isang klinikal na larawan ng Duchenne myodystrophy sa mga babae, ang posibilidad ng isang anomalya sa X chromosome - Shereshevsky-Turner syndrome (XO), Morris syndrome (XY) o mosaicism sa mga sindrom na ito ay dapat munang ibukod.
Ang Duchenne muscular dystrophy, na nagsisimulang umunlad kahit na sa prenatal period, ay mahalagang congenital myopathy at maaaring masuri sa ilang sandali pagkatapos ng kapanganakan sa pamamagitan ng pagsasagawa ng biopsy ng kalamnan at pagtukoy sa aktibidad ng CPK.
Myodystrophy Becker. Kasama ng malubha, malignant na anyo ng X-linked Duchenne myodystrophy, mayroong isang benign form - Becker's disease. Sa mga tuntunin ng mga klinikal na sintomas, ito ay halos kapareho sa Duchenne form, ngunit, bilang isang patakaran, ito ay nagsisimula sa ibang pagkakataon - sa 10-15 taong gulang, malumanay na dumadaloy, ang mga pasyente ay nananatiling magagawang magtrabaho nang mahabang panahon, sa edad na 20. -30 years and later nakakalakad pa sila. Ang pagkamayabong ay hindi nabawasan, kaya ang sakit ay minsan ay sinusubaybayan sa ilang henerasyon ng pamilya: ang isang may sakit na lalaki ay nagpasa ng sakit sa kanyang apo sa pamamagitan ng kanyang anak na babae ("epekto ng lolo"). Ang mga unang sintomas, tulad ng sa Duchenne disease, ay ipinahayag sa pamamagitan ng kahinaan sa mga kalamnan ng pelvic girdle, pagkatapos ay sa proximal lower extremities. Ang mga pasyente ay nagbabago ng kanilang lakad, nakakaranas sila ng kahirapan kapag umakyat sa hagdan, kapag bumabangon mula sa isang mababang upuan. Nailalarawan sa pamamagitan ng pseudohypertrophy ng mga kalamnan ng guya. Ang pagbawi ng calcaneal (Achilles) tendons ay hindi gaanong binibigkas kaysa sa Duchenne disease.
Sa form na ito, walang mga kapansanan sa intelektwal, ang cardiomyopathy ay wala o bahagyang ipinahayag.
Tulad ng ibang X-linked myodystrophies, ang anyo ni Becker ay makabuluhang pinapataas ang aktibidad ng CPK, bagama't sa isang mas mababang lawak kaysa sa Duchenne, hindi hihigit sa 5000 mga yunit. Ang gene para sa Becker's disease, tulad ng Duchenne's disease, ay naisalokal sa maikling braso ng X chromosome; malamang na ang parehong loci ay malapit na nauugnay o allelic. Sa kaibahan sa Duchenne's disease, kung saan halos walang dystrophy, ang abnormal na dystrophy ay synthesized sa Becker's disease. Ang mga pagkakaiba ay matatagpuan din sa biopsy ng kalamnan. Sa muscular dystrophy ni Becker, ang mga fibers ng kalamnan ay karaniwang hindi bilog, ang mga hyaline fibers, katangian ng muscular dystrophy ni Duchenne, ay napakabihirang.
Landouzy-Dejerine myodystrophy (facial-shoulder myodystrophy). Ang sakit ay nakukuha sa autosomal dominant na paraan na may mataas na penetrance ngunit medyo variable expressivity. Ito ay nangyayari nang mas madalas kaysa sa Duchenne myodystrophy (0.4 bawat 100 libong populasyon). Ito ay pinaniniwalaan na ang gene para sa sakit na ito ay naisalokal sa ika-4 na kromosoma. Ang mga babae ay mas madalas magkasakit kaysa sa mga lalaki (3:1), Physical overload, intensive sports, pati na rin ang hindi makatwiran na isinasagawa physiotherapy maaaring mag-ambag sa isang mas malubhang kurso ng sakit.
Ang Landouzy-Dejerine myodystrophy ay isang medyo kanais-nais na kasalukuyang anyo ng muscular pathology. Nagsisimula ito sa edad na mga 20 taon, minsan mamaya. Gayunpaman, sa mga kaso ng pamilya ng sakit, kapag posible na sundin ang dynamics ng mga nakababatang miyembro ng pamilya, posibleng makita ang ilang kahinaan ng mga kalamnan, halimbawa, ang mga kalamnan ng mukha, at sa mas maagang edad. .
Ang kahinaan ng kalamnan at pagkasayang ay unang lumilitaw sa mga kalamnan ng mukha o sinturon sa balikat. Unti-unti, kumakalat ang mga karamdamang ito sa mga kalamnan ng proximal na braso, at pagkatapos ay sa lower limbs. Sa karamihan ng mga kaso, ang mga kalamnan ng nauuna na ibabaw ng mga binti ay unang apektado (na may pag-unlad ng isang nakabitin na paa), pagkatapos ay ang mga kalamnan ng proximal na mga binti. Sa taas ng sakit, ang mga pabilog na kalamnan ng mata at bibig, ang pectoralis major, anterior serratus at mas mababang mga seksyon ng trapezius na kalamnan, ang latissimus dorsi, biceps, triceps na mga kalamnan ng balikat ay lubhang apektado. katangian hitsura mga pasyente: tipikal na mukha myopath na may "transverse smile" ("ngiti ni La Gioconda"), protrusion itaas na labi("tapir lips"), binibigkas ang pterygoid shoulder blades, isang kakaibang deformity ng dibdib na may pagyupi nito sa anteroposterior na direksyon at pag-ikot sa loob ng mga joint ng balikat. Kadalasan mayroong kawalaan ng simetrya ng sugat, kahit na sa loob ng isang kalamnan (hal., orbicularis oculi). Ang pseudohypertrophy ng gastrocnemius, mga deltoid na kalamnan, at kung minsan ang mga kalamnan sa mukha ay maaaring maobserbahan. Ang mga kontrata at pagbawi ay ipinahayag nang katamtaman. tendon reflexes matagal na panahon ay pinananatili, ngunit kung minsan ay bumababa sa maagang yugto.
Ang mga palatandaan ng pinsala sa kalamnan ng puso ay bihira. Ang aktibidad ng serum enzyme ay bahagyang tumaas at maaaring normal. Hindi naghihirap ang talino. Ang pag-asa sa buhay sa karamihan ng mga kaso ay hindi nababawasan. Ang interes ay ang katotohanan na ang EMG sa Landouzy-Dejerine myodystrophy ay kadalasang hindi pangkaraniwan para sa muscular level ng lesyon. Sa ilang mga pasyente (mga miyembro ng parehong pamilya), ang isang pagbawas sa amplitude ng biopotentials, isang uri ng interference ng curve, ay maaaring maobserbahan, sa iba, sa kabaligtaran, isang pagbaba sa dalas at hypersynchronous na aktibidad, kung minsan ay may karaniwang picket. ritmo ng bakod. Dapat itong alalahanin tungkol sa spinal variant, na ginagaya ang Landouzy-Dejerine disease.
Erb-Roth myodystrophy (limb-girdle myodystrophy). Naililipat sa isang autosomal recessive na paraan, ang parehong kasarian ay pantay na apektado. Ang simula ng sakit sa karamihan ng mga kaso ay tumutukoy sa kalagitnaan ng ika-2 dekada ng buhay (14-16 taon), gayunpaman, ito ay inilarawan bilang isang maagang, pseudo-Duchenne form, kapag ang mga unang sintomas ay lumitaw bago ang edad na 10 at ang sakit ay malubha, at isang huli na variant na may simula pagkatapos ng 30 taon.
Ang kurso ng sakit ay maaaring mabilis o mas mabagal, sa karaniwan, ang kumpletong kapansanan ay nangyayari sa loob ng 15-20 taon mula sa simula ng mga unang sintomas. Ang myodystrophy ay nagsisimula alinman sa pinsala sa mga kalamnan ng pelvic girdle at proximal legs (Leiden-Mobius form), o mula sa shoulder girdle (Erb form). Sa ilang mga kaso, ang balikat at pelvic girdle ay apektado nang sabay-sabay. Ang mga kalamnan ng likod at tiyan ay nagdurusa nang malaki. Ang mga pasyente ay may katangian na "pato" na lakad, mahirap bumangon mula sa isang nakahiga at nakaupo na posisyon, binibigyang diin lumbar lordosis. Ang mga kalamnan ng mukha ay hindi apektado sa karamihan ng mga kaso. Para sa form na ito, ang mga contracture at pseudohypertrophy ay hindi karaniwan. Maaaring mangyari ang mga terminal atrophies at tendon retractions. Karaniwang pinapanatili ang katalinuhan. Ang kalamnan ng puso ay halos hindi apektado. Ang antas ng mga enzyme sa serum ng dugo, bilang isang panuntunan, ay nadagdagan, ngunit hindi kasing bilis ng X-linked myodystrophy. May mga indikasyon na sa mga lalaking pasyente ang antas ng CPK ay mas mataas kaysa sa mga babaeng pasyente. Mayroong makabuluhang pagkakaiba sa pagpapahayag ng mutant gene sa iba't ibang miyembro ng pamilya - kasama ang malala klinikal na larawan maaaring mayroong medyo banayad at kahit na nabura ang mga klinikal na sintomas. Karaniwang nangyayari ang kamatayan mula sa mga komplikasyon sa baga.
Dahil ang klinika ng limb-girdle myodystrophy ay lalong handang gayahin ang mga neuromuscular disease ng ibang kalikasan, ito ay kinakailangan, lalo na sa mga sporadic na kaso at may late start sakit, magsagawa ng masinsinang klinikal na pagsusuri upang ibukod ang spinal amyotrophy, polymyositis, metabolic, endocrine, toxic, medicinal, carcinomatous myopathies. Sa nakaraan, nagkaroon ng malinaw na overdiagnosis ng ganitong uri ng muscular dystrophy.
Paggamot ng muscular dystrophies. Ang mga opsyon sa therapeutic para sa muscular dystrophies ay napakalimitado. Etiological at pathogenetic na paggamot halos wala. Ang symptomatic na paggamot ay pangunahing naglalayong pigilan ang pag-unlad ng contractures, pagpapanatili ng umiiral na lakas ng kalamnan at, marahil, sa ilang pagbawas sa rate ng pagkasayang. Ang pangunahing gawain ay upang i-maximize ang panahon kung saan ang pasyente ay nakakagalaw nang nakapag-iisa, dahil ang mga contracture, scoliosis, at mga sakit sa paghinga ay mabilis na tumataas sa posisyong nakahiga. Medical complex dapat isama ang mga therapeutic exercise, masahe, orthopaedic measures, drug therapy.
Ang therapeutic gymnastics ay binubuo ng mga passive at aktibong paggalaw na ginagawa sa lahat ng mga joints sa iba't ibang posisyon: nakatayo, nakaupo, nakahiga, na may iba't ibang posisyon ng mga limbs. aktibong paggalaw Mas mainam na gumanap sa isometric mode. Ang himnastiko ay dapat gawin nang regular nang maraming beses sa isang araw. Kasabay nito, ang isa ay dapat na bigyan ng babala laban sa labis na ehersisyo, lalo na ang mga sinamahan ng overstretching ng mga kalamnan. Mahalaga (lalo na pagkatapos ng immobilization ng pasyente) ay mga pagsasanay sa paghinga.
Mga aktibidad sa orthopedic konserbatibo (espesyal na splints) at pagpapatakbo kalikasan (achillotomy, transection ng gastrocnemius kalamnan), na naglalayong iwasto contractures at umuusbong na pathological limbs setting, din layunin upang mapanatili ang posibilidad ng malayang paggalaw. Sa bawat kaso, kinakailangang indibidwal na timbangin ang inaasahang benepisyo at posibleng pinsala mula sa surgical intervention. Dapat itong isipin na madalas (sa partikular, na may matinding hyperlordosis at kahinaan ng quadriceps femoris na kalamnan), ang posisyon ng equinovarus ng mga paa ay may katumbas na kahalagahan, at pagkatapos, halimbawa, achillotomy, ang pasyente ay maaaring ganap na immobilized. Sa pagbuo ng contractures, inirerekumenda na maingat na iunat ang mga kalamnan hanggang 20-30 beses sa isang araw, na sinusundan ng splinting sa panahon ng pagtulog.
Ang therapy sa droga ay nagsasangkot ng appointment ng mga metabolic na gamot na naglalayong punan ang kakulangan ng enerhiya at protina, ngunit ang kanilang pagiging epektibo ay napaka-duda. Ginagamit ang mga antagonist ng calcium (dahil sa isang depektong natukoy sa sakit na Duchenne mga lamad ng cell humahantong sa pagtaas ng paggamit ng calcium sa cell), immunomodulators, phosphorus-containing compounds (ATP, phosphaden), bitamina E (100 mg pasalita 3 beses sa isang araw). Ipinakita na ang paggamit ng prednisolone (0.75 mg/kg bawat araw) sa sakit na Duchenne ay maaaring kapansin-pansing magpapataas ng lakas ng kalamnan, ngunit ang epektong ito ay nagpapatuloy nang hindi hihigit sa isang taon at sa pangkalahatan ay hindi nakakaapekto sa kinalabasan ng sakit. Dahil sa seryoso side effects, nagmumula vri pangmatagalang paggamit gamot, ang paggamit nito ay hindi nararapat. Mga pagtatantya ng epekto mga anabolic steroid kontrobersyal at ang kanilang appointment ay madalas na nauugnay sa hindi makatarungang panganib. Kapag sinusuri ang epekto ng ilang mga gamot sa Duchenne, dapat tandaan na may katamtamang kalubhaan ng sakit sa mga pasyente na may edad na 3-6 na taon, maaaring mayroong isang kamag-anak na pag-stabilize ng estado na nauugnay sa pag-unlad na nauugnay sa edad ng muscular system, ang pagkuha ng mga kasanayan sa motor, na maaaring pansamantalang magbayad para sa patuloy na dystrophic na proseso.
Ang tiyak na kahalagahan ay ang pagwawasto ng nutrisyon ng pasyente, ang isang diyeta na mataas sa protina at mababa sa taba at nabawasan sa mga calorie na may pinakamainam na nilalaman ng mga bitamina at microelement ay inirerekomenda. Ang isang mahalagang papel ay ginampanan ng sikolohikal na suporta ng pasyente, ang pagpapatuloy ng edukasyon, at ang tamang propesyonal na oryentasyon.
Pahina 44 ng 44
Ang mga kalamnan ng kalansay ay kasangkot sa proseso ng pathological na may iba't ibang degenerative, metabolic at nagpapaalab na sakit. Sa karamihan ng mga kaso, nagreresulta ito sa pagkabulok ng mga fibers ng kalamnan, at sa mga talamak na anyo, ang kanilang kapalit. nag-uugnay na tisyu at mataba. Ang mga proximal na grupo ng kalamnan ay mas nasira kaysa sa mga distal, pati na rin ang mas mababang mga paa na may kaugnayan sa mga nasa itaas. Ang isang may sakit na bata ay nakikilala sa pamamagitan ng tinatawag na duck (waddling) gait, hindi makatakbo, umakyat sa hagdan at bumangon kung siya ay nasa posisyong nakaupo. Ang kanyang mga tendon reflexes ay nalulumbay, ang antas ng kanilang pagkalipol ay proporsyonal sa antas ng pagpapahina ng lakas ng kalamnan. Hindi apektado ang pagiging sensitibo.
Kasama sa mga diagnostic na mahalagang pamamaraan ng laboratoryo ang pagpapasiya ng aktibidad ng mga enzyme, lalo na ang creatine phosphokinase, sa suwero. Ang enzyme na ito, na nagpapagana ng reaksyon: phosphocreatine + ADP-creatine + ATP, ay naroroon pangunahin sa mga selula ng utak at tissue ng kalamnan. Sa ilang nagkakalat na sakit sa kalamnan, lalo na ang muscular dystrophy, ang mga labis na halaga nito ay tumagos sa intercellular space at dugo. Sa mga pasyente, ang aktibidad ng serum lactate dehydrogenase at glutamine oxaloacetic transaminase ay karaniwang tumataas, ngunit ang kanilang malawak na pamamahagi sa iba pang mga tisyu, kabilang ang atay, ay binabawasan ang pagtitiyak ng pagsubok. Karaniwan, ang isang biopsy ng tissue ng kalamnan ay kinakailangan upang linawin ang diagnosis.
Mga nagpapaalab na sakit sa kalamnan. Ang pamamaga ng tissue ng kalamnan ay kasama ng ilang mga impeksyon, lalo na ang trichinosis, toxoplasmosis, at ang mga sanhi ng Coxsackie virus. Kadalasan ito ay bahagi ng mga sakit sa collagen, kabilang ang dermatomyositis, lupus erythematosus, periarteritis nodosa, at rheumatoid arthritis.
Polymyositis. Ang nagkakalat na nakahiwalay na pamamaga ng kalamnan ng hindi kilalang etiology ay tinatawag na polymyositis. Ito ay nailalarawan sa pamamagitan ng isang mabilis na progresibong kurso, kahinaan at sakit sa proximal na mga grupo ng kalamnan. Kadalasan, ang mga kalamnan ng leeg ay kasangkot sa proseso, at samakatuwid ay nagiging mahirap para sa bata na itaas ang kanyang ulo at panatilihin ito sa posisyon na ito. Ang mga palatandaan sa laboratoryo ng pamamaga ng kalamnan ay kinabibilangan ng pagtaas ng ESR at ang bilang ng mga leukocytes. Gayunpaman, ang kanilang kawalan ay hindi nagbubukod ng polymyositis. Ang mga antas ng serum enzyme ay karaniwang nakataas. Sa biopsy ng kalamnan, ang pagkabulok at bahagyang pagbabagong-buhay ng mga hibla at ang kanilang paglusot ng mga selulang lymphoid ay tinutukoy. Mahirap ibahin ang polymyositis mula sa muscular dystrophy at dermatomyositis. Maaaring ito ay kumakatawan sa isang hindi tipikal na anyo ng dermatomyositis, bagaman ang histolohiya ng dalawang kondisyon ay medyo naiiba: ang dermatomyositis ay nailalarawan sa pamamagitan ng vasculitis, na kadalasang wala sa polymyositis. Ang pagbabala para sa huli ay medyo mas kanais-nais. Ang paggamot na may corticosteroids ay sinamahan ng isang epekto, ngunit kapag kinansela ang mga ito, maaaring mangyari ang pagbabalik sa dati.
Progressive ossifying myositis. Ang etiology ng bihirang connective tissue at sakit sa kalamnan na ito ay hindi alam. Iniulat na ang mga kapatid, kabilang ang kambal, ay nagdurusa dito, at ito ay naililipat sa mga kadugo sa isang direktang linya. Ito ay pinaniniwalaan na ang sakit ay minana sa isang autosomal dominant na paraan. Ang mga lalaki ay nagkakasakit ng 2-3 beses na mas madalas kaysa sa mga babae.
Ang mga palatandaan ng pathological ay nakasalalay sa yugto ng sakit. Sa mga unang yugto, ang lokal na edema at nagpapasiklab na mga cell infiltrates ay matatagpuan sa mga kalamnan at tendon. Nang maglaon, ang mga lugar ng pamamaga ay pinalitan ng granulation tissue, at kalaunan ay nabubuo ang mga bahagi ng cartilage at bone tissue sa mga sugat.
Halos 75% ng mga may sakit na bata ay mayroon Problema sa panganganak pag-unlad, kadalasang hindi pag-unlad ng mga daliri at ankylosis ng mga phalanges ng mga unang daliri at hindi pag-unlad ng mga unang daliri, polydactyly, kurbada ng mga daliri, syndactyly (binti), deformity auricle pagkabingi, nawawalang ngipin. Ang parehong congenital malformations ay maaaring nasa mga kamag-anak ng pasyente na hindi nakabuo ng progresibong sakit ng connective tissue at mga kalamnan. Ang edad kung saan maaaring magsimula ang myositis ossificans ay nag-iiba mula sa kapanganakan hanggang sa mas matandang pagkabata. Karaniwan, tatlong yugto ang nakikilala: 1) limitado, kadalasang mainit at malambot sa pagpindot, lumilitaw ang mga malagkit na pamamaga ng malambot na tisyu sa mga lugar ng menor de edad na lokal na pinsala; 2) pagkatapos ng ilang araw, nawawala ang mga sintomas ng pamamaga, at tumigas ang sugat; 3) nangyayari ang ossification ng apektadong lugar. Paminsan-minsan, lumilitaw ang mga bagong sugat, pangunahin sa leeg at likod. pangunahing sintomas torticollis ay maaaring maging kung ang proseso ay nabuo sa sternocleidomastoid na kalamnan. Sa kalaunan, ang ossification ay umaabot sa maraming tendon at ligaments. Ang ankylosis ng gulugod at mga kasukasuan ng mga braso at binti ay makikita (Larawan 21-5). Ang pamamaga ay maaaring kumalat sa temporomandibular joints, na nagpapahirap sa paggalaw ng pagnguya. Ang mga buto ay maaaring lumabas sa balat. Sa pagbibinata, ang sakit ay madalas na humahantong sa kumpletong immobilization at kamatayan dahil sa pagkabigo sa paghinga at paghinto ng paghinga, bagama't may mga ulat ng mga kaso ng kaligtasan. Sa ossifying myositis, may mataas na panganib na magkaroon ng osteogenic sarcoma.
kanin. 21-5. Batang may progresibong myositis ossificans (karaniwang postura na may paninigas ng leeg at likod).
Minsan ang proseso ng pathological ay limitado sa site ng isang nakaraang pinsala sa malambot na tissue (miositis ossificans circumscripta). Ang malawakang pag-calcification ng tissue ng kalamnan ay maaari ding mangyari sa talamak na polymyositis at dermatomyositis.
resulta mga pamamaraan sa laboratoryo walang diagnostic value ang mga pag-aaral.
Ang mga antas ng serum ng calcium, phosphorus, alkaline phosphatase, pati na rin ang aktibidad ng creatine phosphokinase at iba pang mga enzyme ay nananatiling normal. buto sa pokus ng pinsala ay hindi naiiba sa istraktura mula sa pamantayan.
Mga Umiiral na Pamamaraan hindi kasiya-siya ang mga paggamot. Sa ilang mga kaso, ang paghina sa pag-unlad ng sakit ay napansin sa paggamit ng ACTH at iba pang corticosteroids. Ang kanilang papel sa huling resulta ng paggamot ay kaduda-dudang.
Endocrine at metabolic myopathies. Ang myopathy sa hyperthyroidism ay isang medyo bihirang komplikasyon. Ito ay nailalarawan sa pamamagitan ng ptosis, bilateral paresis ng facial muscles at muscles ng proximal limbs. Kasabay nito, ang ilang mga sintomas ng hyperthyroidism ay maaaring natatakpan ng kahinaan ng kalamnan, ngunit ang tachycardia, pagtaas ng pagpapawis at pagtaas thyroid gland. Ang mga tendon reflexes, hindi tulad ng maraming iba pang anyo ng myopathy, ay nananatiling normal. Pagkatapos ng pagwawasto ng hyperthyroidism, ang kahinaan ng kalamnan ay unti-unting nawawala.
Myopathy sa hypothyroidism. Ang hypothyroidism sa mga sanggol ay maaaring nauugnay sa kahinaan ng kalamnan at hypotension. Sa mas matatandang mga bata na may myxedema, ang mga contraction ng kalamnan at pagpapahinga ay bumagal, sa ilang mga kaso ang hypertrophy ng kalamnan ay nabanggit (Debre-Semelen syndrome). Ang kumbinasyon ng mga palatandaan tulad ng kahinaan ng kalamnan at hypertrophy ay nagpapahiwatig ng muscular dystrophy.
Myopathy sa panahon ng paggamot na may corticosteroids. Maaari nitong gawing kumplikado ang sakit na Itsenko-Cushing, ngunit mas madalas na nabubuo sa paggamot ng malalaking dosis ng mga sintetikong steroid. Ang kahinaan ay lalong kapansin-pansin sa mga kalamnan ng pelvic girdle, na nagpapakita ng sarili sa isang waddling (duck) gait, kahirapan sa pag-akyat sa hagdan at sinusubukang bumangon mula sa isang posisyon sa pag-upo. Wala ang knee jerk. Maaaring mangyari ang pagnipis ng kalamnan. Ang mga myopathic na pagbabago sa tissue ng kalamnan ay kadalasang hindi gaanong mahalaga kahit na may matinding kahinaan. lakas ng kalamnan pagkatapos ng pagpawi ng corticosteroids, ito ay dahan-dahang bumabawi (sa loob ng ilang buwan).
Myopathy sa hyperparathyroidism. Ang hyperparathyroidism ay maaaring nauugnay sa kahinaan at hyporeflexia dahil sa hyperkalemia. Kadalasan ay mabilis silang nawawala pagkatapos ng parathyroidectomy.
Ang kakulangan ng carnitine (lipid myopathy) ay sinamahan ng akumulasyon ng malalaking halaga ng mga lipid sa mga kalamnan at, bilang isang resulta, ang paglabag sa supply ng enerhiya ng huli. Ang carnitine ay isa sa mga mahahalagang bahagi ng system na nagsisiguro sa paglipat ng mga fatty acid mula sa mahabang kadena mula sa cytosol hanggang sa mitochondria, kung saan sila ay sumasailalim sa oksihenasyon. Ang kahinaan ng kalamnan ay bubuo sa dalawang anyo ng kakulangan sa carnitine.
Ang kakulangan ng carnitine sa mga kalamnan ay klinikal na kinakatawan ng progresibong kahinaan ng kanilang mga proximal na grupo, mas madalas sa mga mag-aaral at kabataan. Minsan ang kahinaan ay paulit-ulit at sinamahan ng myoglobinuria. Sa malalang kaso, maaaring mangyari ang paralisis ng mga kalamnan sa paghinga. Ang mga antas ng serum ng enzymes (creatine kinase at aldolase) ay tumaas. Ang electromyogram ay nagpapakita ng mga hindi tiyak na pagbabago na katangian ng myopathy. Sa biopsy ng kalamnan, makikita mo ang isang malaking bilang ng mga droplet ng taba. Ang antas ng carnitine sa suwero ay hindi nagbabago, ngunit sa mga kalamnan ay bumababa ito. Ang pagkilala sa patolohiya ay mahalaga, dahil maaari itong gamutin. Madalas itong napagkakamalang muscular dystrophy. Ang epekto ay maaaring mangyari pagkatapos ng oral administration ng 100 mg / (kg / day) carnitine. Sa ilang mga kaso, ang paggamot na may corticosteroids ay epektibo.
- Ang systemic carnitine deficiency ay ipinakikita ng progresibong myopathy, kabilang ang cardiomyopathy, at liver dysfunction, na sinamahan ng isang klinika ng hepatic encephalopathy tulad ng Reye's syndrome. Ang kakulangan ng carnitine ay naiiba mula sa huli sa paulit-ulit na kurso nito at binibigkas ang kahinaan ng kalamnan na nagpapatuloy sa pagitan ng mga panahon ng exacerbation ng encephalopathy. Ang antas ng creatine phosphokinase sa suwero ay kapansin-pansing tumaas, ang dami ng carnitine ay nabawasan kapwa sa suwero at sa mga kalamnan. Ang mga pagbabago sa biopsy ay katulad ng mga may kakulangan sa carnitine sa tissue ng kalamnan. Ang mga katulad na klinikal at morphological na pagbabago, kabilang ang carnitine deficiency, ay maaaring makita sa paglabag sa metabolismo ng mga organic acids, halimbawa, sa methylmalonic at glutaric aciduria (secondary carnitine deficiency).
kanin. 21-6. Isang bata na may congenital na kawalan ng kaliwang pectoralis major na kalamnan. 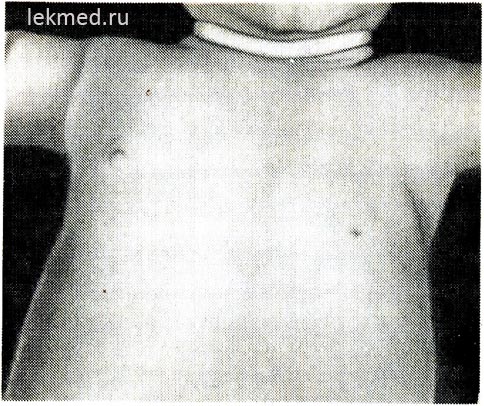
Pansinin ang kawalan ng anterior axillary fold at ang low-lying nipple.
Ang paggamot ay binubuo sa pagpapanatili ng pasyente sa isang diyeta na mayaman sa carbohydrates at mababang taba, at pagkuha ng carnitine sa pang-araw-araw na dosis na 100 mg / kg.
Mga depekto sa congenital na kalamnan. Congenital na kawalan ng kalamnan. Ang kakulangan sa pag-unlad ng kalamnan ay maaaring maging pangkaraniwan at humantong sa kumpletong pagbara ng magkasanib na paggalaw o congenital arthrogryposis. Bilang isang depekto sa kapanganakan, ang isang kalamnan ay madalas na nawawala. Ang isang medyo karaniwang anomalya ay ang kawalan ng sternal na bahagi ng pectoralis major na kalamnan (Larawan 21-6), sa ilang mga kaso ang depektong ito ay pinagsama sa syndactyly sa apektadong bahagi (Poland's syndrome). Ang kawalan ng pectoral na kalamnan ay madalas na kasama ng muscular dystrophy. Ang kawalan ng congenital ng mga kalamnan ng tiyan ng tiyan ay kadalasang nauugnay sa mga depekto sa pag-unlad ng daanan ng ihi.
kanin. 21-7. Neck deformity at facial asymmetry sa isang batang lalaki na may congenital torticollis, na hindi ginagamot mula noong edad na 12. 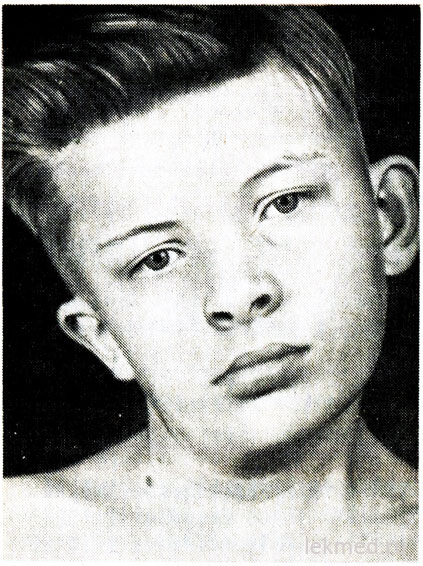
Ang congenital torticollis ay sanhi ng unilateral shortening o contracture ng sternocleidomastoid muscle. Ang ulo ng pasyente ay nakatagilid patungo sa contracture, at ang baba ay nakadirekta pababa sa tapat na direksyon (Larawan 21-7). Kapag sinusubukang itama ang posisyon ng ulo, nararamdaman ang makabuluhang paglaban ng kalamnan. Sa apektadong kalamnan, ang mga lugar ng compaction ay palpated. Ang sanhi ng depekto ay hindi malinaw, sa loob ng mahabang panahon ito ay itinuturing na resulta ng pinsala sa panganganak. Gayunpaman, ang torticollis ay nangyayari sa mga batang ipinanganak sa pamamagitan ng operasyon. caesarean section; ito ay nagpapahiwatig na sa ilang mga kaso ang sanhi ng depekto ay tumutukoy sa intrauterine period. Ang Torticollis ay dapat na naiiba mula sa pathological head tilt dahil sa deformity ng cervical vertebrae, tulad ng Klippel-Weil anomaly, at mula sa mga bali o dislokasyon ng cervical vertebrae. Ang mga ito ay hindi kasama ng pagsusuri sa X-ray. Sa mas matatandang mga bata, ang pagkiling ng ulo ay maaaring dahil sa strabismus, dystonia, mga tumor ng posterior cranial fossa at servikal spinal cord, myositis ossificans, cervical lymphadenitis o diaphragmatic hernia. Sa karamihan ng mga kaso, maaaring maitama ang congenital torticollis therapeutic gymnastics. Gayunpaman, kapag talamak na anyo torticollis ay nagreresulta sa isang asymmetrical development ng mukha at ulo (tingnan ang Fig. 21-7), na maaaring mangailangan ng dissection ng kalamnan para sa mga layuning kosmetiko.
congenital myopathies. Kasama sa grupong ito ang ilang mga bihirang uri ng namamana na sakit kung saan lumalabas ang kahinaan ng kalamnan at hypotonia mula sa pagkabata (tingnan ang Talahanayan 22-1). Ang kanilang tumpak na diagnosis ay pinakamahalaga sa mga tuntunin ng pagtataya. Sa pangkalahatan, ito ay kanais-nais para sa normal na aktibidad sa buhay at pag-asa sa buhay, sa kaibahan sa sakit na Werdnig-Hoffmann o congenital muscular dystrophy. Karaniwang nakakatulong ang biopsy ng kalamnan upang matukoy ang mga congenital myopathies.
- Sakit ng gitnang core. Ang gitnang bahagi ng mga fibers ng kalamnan ay may kulay na abnormal, ngunit pare-pareho. Ang isang electron microscopic na pagsusuri ay nagpapakita ng pagbaba sa bilang ng mitochondria at isang pag-ubos ng sarcoplasmic reticulum sa gitnang bahagi ng mga hibla.
Nemaline myopathy. Ang terminong "non-crimson" ay ipinaliwanag sa pamamagitan ng katotohanan na ang mga istraktura na tulad ng sinulid ay tinutukoy sa mga fibers ng kalamnan.
kanin. 21-8. Myotonic contraction ng dila (a) na may isang matalim na suntok na may percussion hammer sa kanang kalahati nito at eyelids (b) sa isang bata na may hyperkalemic form ng familial periodic paralysis. 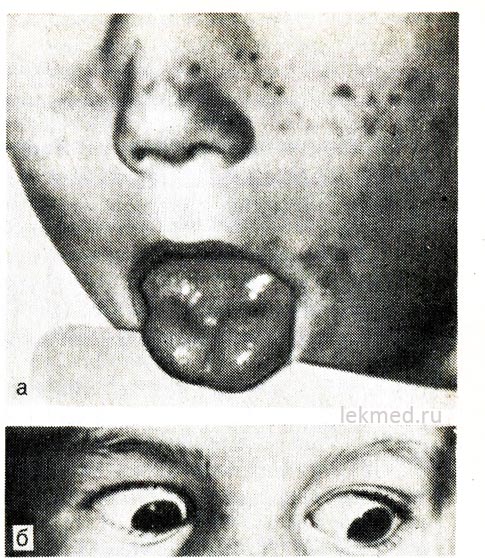
Kapag nakatingin sa ibaba, ang talukap ng mata ay nananatiling nakakontrata.
Ipinapahiwatig ng data ng pagsusuri ng mikroskopiko ng elektron na ito ang resulta ng mga pagbabago sa mga Z-band ng myofibrils.
Mitochondrial myopathies. Ang ilang mga anyo ng myopathies ay naiulat kung saan ang pinakamahalagang pagbabago ay nangyayari sa mitochondria ng mga fibers ng kalamnan. Maaari silang kapansin-pansing tumaas kapwa sa bilang at sa laki. Ang kahinaan ng kalamnan at hypotonia ay maaaring matukoy na sa pagkabata, ngunit kung minsan ay kapansin-pansing umuunlad lamang sa paaralan. Ang cardiomyopathy, encephalopathy at lactic acidemia ay kadalasang sinasamahan ng grupong ito ng myopathies.
Myotonia. Ang kundisyong ito ay isang tanda ng iba't ibang sakit sa kalamnan, tulad ng myotonia dystrophica, hyperkalemic familial paroxysmal palsy, at glycogen storage disease. Ang myotonia ay tinukoy bilang isang makabuluhang pagkaantala sa pagpapahinga ng kalamnan pagkatapos ng boluntaryo o sapilitang mga contraction. Sa klinika, ito ay nagpapakita ng sarili sa kawalan ng kakayahang alisin ang kamao o sa nakikitang matagal na pag-urong ng mga kalamnan kasunod ng kanilang pagpapasigla, na ipinahayag sa isang matalim na pangangati (Larawan 21-8). Ito ay mapapansin kung natamaan mo ang isang mababaw na grupo ng mga kalamnan gamit ang isang percussion hammer, halimbawa, ang mga kalamnan ng dila o ang palmar surface sa lugar ng elevation ng unang daliri. Ang Myotonia ay kinumpirma ng data ng electromyography. Sa kasong ito, ang katangian ng kusang aktibidad ng mga kalamnan ay kapansin-pansin pagkatapos ng kanilang pagpapahinga o boluntaryong pag-urong (myotonic discharges).
Myotonia congenita (sakit ni Thomsen). Ang tanging palatandaan ng sakit na ito, na minana ng dominanteng uri, ay myotonia. Maaari itong magpakita mismo sa pagkabata sa anyo ng isang pagbagal sa mga paggalaw ng paglunok at pagsusuka
epekto ng kawalan ng kakayahan sa normal na pagpapahinga ng mga kalamnan ng pharynx. Sa mas matanda pagkabata Ang myotonia ay nagpapakita ng sarili bilang ang kawalan ng kakayahan ng pasyente na i-unclench ang kanyang mga daliri na nakakuyom sa isang kamao. Sa unang pagtatangka na magsagawa ng ilang uri ng paggalaw, ang mga kalamnan ng bata ay nagiging matigas. Sa paulit-ulit na pag-uulit ng parehong paggalaw, medyo nakakarelaks sila. Kaya, halimbawa, ang isang may sakit na bata ay nakakaranas ng malaking paghihirap sa simula ng pagkilos ng paglalakad. Karaniwang ginagawa niya ang mga unang hakbang nang mabagal at mabagal. Pagkatapos ng ilang segundo, nagiging normal o halos normal ang lakad. Ang mga sintomas ng myotonia ay pinalala ng hindi kanais-nais na emosyonal na kalagayan ng pasyente at paglamig ng katawan. Ang lakas ng kalamnan ay nananatiling normal, ang mga kalamnan ay sapat na binuo at kadalasang kapansin-pansing pinalaki, na lumilikha ng maling impresyon sa konstitusyon ng atletiko ng pasyente.
Ang diagnosis ay batay sa mga klinikal na natuklasan at data ng electromyography. Ang aktibidad ng serum enzyme ay nasa loob ng normal na mga limitasyon. Ang tanging histological sign ay hypertrophy ng mga fibers ng kalamnan.
Ang sakit ay naiiba mula sa dystrophic myotonia sa pamamagitan ng kawalan ng kalamnan kahinaan at pagkasayang at dystrophic pagbabago sa kalamnan tissue biopsy. Ang paggamot na may novocaine o quinidine sulfate ay sinamahan ng isang epekto at ipinahiwatig para sa mga functional disorder. Ang kurso ng sakit ay karaniwang benign, at ang kondisyon ng pasyente ay maaaring mapabuti sa edad.
paroxysmal paralysis. Ang pangkat ng mga sakit na ito ay nailalarawan sa pamamagitan ng panaka-nakang kahinaan ng kalamnan na may kumpleto o halos kumpletong pagpapanumbalik ng lakas ng kalamnan sa pagitan ng mga pag-atake. Kasama rin dito ang kakulangan ng muscle phosphorylase (McArdle's disease).
Hyperkalemic paroxysmal paralysis. Ang hereditary episodic adynamia, o paramyotonia, ay naipapasa ayon sa dominanteng uri at lalong malala sa mga lalaki. Ito ay karaniwang nagsisimula sa maagang pagkabata (minsan sa pagkabata). Nangyayari ang mga pag-atake sa panahon ng pahinga pagkatapos ng mabigat na pagkarga ng kalamnan. Mabilis na umuunlad ang kahinaan at maaaring tumagal ng ilang oras. Ito ay lalo na nadama sa mga binti; Ang paggana ng paghinga ay karaniwang hindi napinsala. Kadalasan ang adynamia ay sinamahan ng myotonia, na nagpapatuloy sa pagitan ng mga pag-atake, na kung saan ay pinaka-malinaw na ipinahayag sa anyo ng isang pagkaantala sa paggalaw ng mga eyelid kapag tumitingin pababa (tingnan ang Fig. 21-8, b).
Ang mga antas ng serum potassium ay madalas na nakataas sa panahon ng isang pag-atake, ngunit maraming pag-aaral sa panahon ng ilang mga pag-atake ay maaaring kailanganin upang matukoy ito nang may katiyakan. Posible na artipisyal na pukawin ang isang pag-atake sa tulong ng isang potassium load (2-3 g pasalita), ngunit dapat itong isagawa lamang sa ilalim ng kontrol ng ECG. Ang paulit-ulit na pag-atake ay pinipigilan ng diacarb. Ang mga malubhang anyo ng sakit ay nailalarawan sa pamamagitan ng pag-unlad ng talamak, banayad na kahinaan at degenerative na pagbabago sa mga kalamnan.
Hypokalemic paroxysmal paralysis. Ang paroxysmal paralysis ng pamilya, na minana din ayon sa dominanteng uri, ay lalong mahirap sa mga lalaki. Sa kaibahan sa hyperkalemic form, ang unang pag-atake ay lumilitaw sa huling bahagi ng pagkabata o maagang pagbibinata. Ang dahilan ay ang pagkain ng masaganang pagkain na mayaman sa carbohydrates, o pagpapahinga pagkatapos pisikal na Aktibidad. Ang pag-atake ay karaniwang nagsisimula sa sa susunod na umaga pagkatapos ng mabigat na pisikal na pagsusumikap at isang mabigat na pagkain. Ito ay nailalarawan sa pamamagitan ng kahinaan ng kalamnan at areflexia. Maaaring may kapansanan ang paggana ng paghinga. Maaaring sumali ang arrhythmia, kabilang ang ventricular extrasystole at tachycardia. Ang mga pag-atake ay maaaring tumagal ng higit sa 24 na oras. Sa paralytic phase, ang antas ng potasa sa serum ay karaniwang bumababa (2-3 mmol / l). Ang pinagbabatayan na depekto ay hindi alam. Ang mga pasyente na may paulit-ulit na matinding pag-atake ay nagkakaroon ng talamak na panghihina ng kalamnan at mga pagbabago sa pathological sa mga kalamnan. Ang paggamot sa panahon ng pag-atake ay binubuo ng pagkuha ng potassium chloride; ang paunang dosis nito ay 2-3 g. Nakakatulong ang Diakarb na bawasan ang dalas ng mga seizure.
Paroxysmal myoglobinuria (idiopathic myoglobinuria). Ang idiopathic myoglobinuria ay isang magkakaibang grupo ng mga karamdaman kung saan ang mga pag-atake ng paralisis na may myoglobinuria ay kusang nangyayari o pagkatapos ng matinding ehersisyo. Ang sakit ay minana sa isang nangingibabaw na paraan, na naka-link sa X chromosome. Ang mga kalamnan, kadalasan ang mga binti at hita, ay nagiging masakit at namamaga sa panahon ng pag-atake. Ang ihi ay nagiging madilim na pula o Kulay kayumanggi. Ang myoglobinuria ay maaaring magdulot ng renal tubular necrosis, na nakamamatay dahil sa pagkabigo sa bato.
Ang diagnosis ay nakumpirma sa pamamagitan ng pagtuklas ng myoglobulin sa ihi. Ang isang positibong pagsusuri sa benzidine sa kawalan ng mga erythrocytes sa ihi ay nagpapatunay sa pagkakaroon ng myoglobin dito, lalo na kung ang hemoglobin ay hindi napansin sa suwero. Ang hemoglobin ay tinutukoy ng spectrophotometry. Ang paroxysmal myoglobinuria ay dapat na makilala mula sa McArdle disease, carnitine palmityltransferase deficiency, at myoglobinuria kasunod ng hindi pangkaraniwang matinding ehersisyo o pinsala sa kalamnan sa malusog na tao. Ang myoglobinuria pagkatapos ng mabigat na pag-eehersisyo ng kalamnan ay nangyayari sa pseudohypertrophic muscular dystrophy (Duchenne disease).
Ang paggamot ay binubuo ng bed rest; isagawa kung kinakailangan artipisyal na bentilasyon baga. Upang maiwasan ang pagkabigo sa bato, kinakailangang magreseta ng maraming inumin sa pasyente.
Kakulangan ng carnitine palmityltransferase. Sa kakulangan ng enzyme na ito, ang paglipat ng mga long-chain fatty acid sa mga segment ng mitochondrial, kung saan isinasagawa ang oksihenasyon at paggawa ng mga ketone, ay nagambala. Ang type II isoenzyme deficiency ay minana sa isang recessive na paraan. Dahil sa kakulangan nito, ang ketogenesis sa mga tisyu, kabilang ang kalamnan at atay, ay nagambala. Ang mga unang palatandaan ng sakit ay lumilitaw nang mas madalas sa mga bata sa paaralan at pagbibinata. Binubuo ang mga ito ng paulit-ulit na yugto ng pananakit ng kalamnan, panghihina, at lagnat pagkatapos ng ehersisyo o pag-aayuno. Ang myoglobinuria na kasama ng mga seizure ay maaaring humantong sa pagkabigo sa bato. Ang pag-aayuno ay humahantong sa hypoglycemia. Sa pagitan ng mga pag-atake, mukhang malusog ang mga bata. Ang sakit ay dapat na naiiba mula sa iba pang mga kondisyon na sinamahan ng panaka-nakang kahinaan at myoglobinuria. Ang pamamaraan para sa pagtukoy ng aktibidad ng carnitine palmityl transferase ay may kaugalian na diagnostic na halaga. Ito ay bumababa sa kalamnan at mga tisyu ng atay, leukocytes at fibroblast culture. Ang pagkain ng carbohydrate-rich, low-fat diet ay maaaring makatulong na mabawasan ang mga seizure.
Muscular dystrophies. Ang mga anomalyang ito ay nabibilang sa isang pangkat ng mga sakit sa pamilya na sinamahan ng pagkabulok ng mga fibers ng kalamnan. Ang pag-uuri ng muscular dystrophies ay batay sa mga tampok tulad ng oras ng pagsisimula, bilis ng pag-unlad, pamamahagi ng mga sugat ayon sa grupo ng kalamnan, at paraan ng pagmamana.
Pseudohypertrophic muscular dystrophy. Ang pagkabata, o ang Duchenne form, ay ang pinakakaraniwang anyo ng muscular dystrophy; ang dalas nito ay 0.14 bawat 1000 bata. Sa klasikal na anyo, ito ay nangyayari lamang sa mga lalaki, at ang pamana na nauugnay sa X chromosome ay nangyayari sa humigit-kumulang 50% ng mga proband. Sa ibang mga kaso, ang sakit ay dahil sa mga bagong mutasyon. Ang isang bihirang anyo ng muscular dystrophy ay iniulat, na klinikal na magkapareho sa anyo ng Duchenne, ngunit minana ng isang recessive na uri na may parehong dalas ng sakit sa mga lalaki at babae. Ang mapagkakatiwalaang pag-diagnose ng sakit ay bihirang posible sa isang batang wala pang 3 taong gulang. Ang kasaysayan ay karaniwang nagpapahiwatig na ang bata ay naantala ang pag-unlad mga function ng motor, nagsimula siyang umupo, maglakad at tumakbo nang huli, na, siyempre, ay nagpapahiwatig ng higit pa maagang simula mga sakit. Waddling (duck) gait, kahirapan sa pag-akyat sa hagdan, calf muscle hypertrophy ay karaniwan. mga klinikal na pagpapakita. Sa ilang mga kaso, ang ibang mga kalamnan ay kasangkot din sa proseso, lalo na ang deltoid, brachioradialis, at mga kalamnan ng dila.
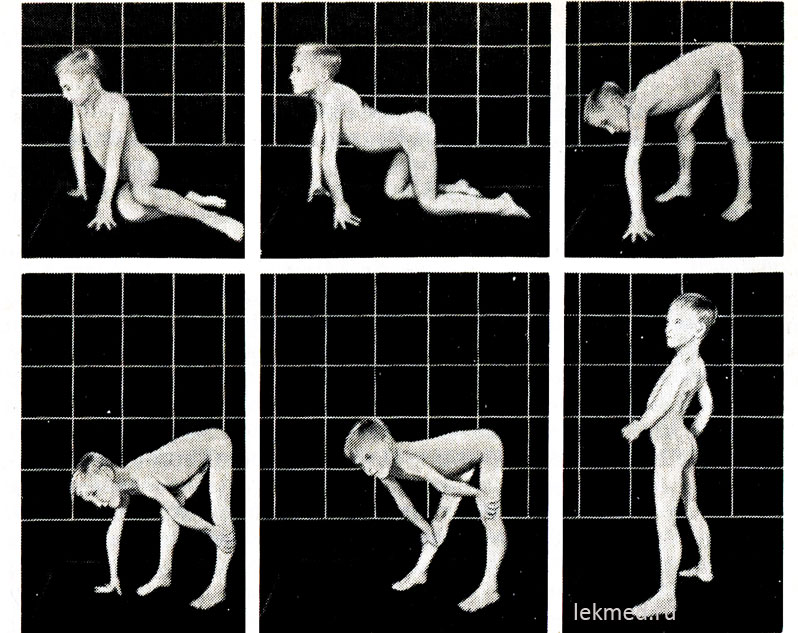
kanin. 21-9. Mga karaniwang postura kapag bumangon mula sa sahig (Govers sign) sa isang 7 taong gulang na bata na may pseudohypertrophic myopathy.
Sa nakatayong posisyon (huling larawan), mayroong isang makabuluhang binibigkas na lordosis.
Sa simula ng sakit, ang mga hypertrophied na kalamnan ay may makabuluhang lakas, ngunit sa paglaon ay bumababa ito (pseudohypertrophy), dahil ang pagtaas sa mass ng kalamnan ay nangyayari dahil sa kanilang mataba na pagpasok. Ang lakas ng hypertrophied gastrocnemius na kalamnan ay makabuluhang lumampas sa lakas ng mga kalamnan ng nauunang ibabaw ng binti, na nagpapaliwanag ng madalas na mga contracture ng calcaneal tendon at ang bata na naglalakad sa kanyang mga daliri. Ang kahinaan ng mga kalamnan ng pelvic girdle ay ipinahayag sa isang katangian na lakad ng pato (panginoon) at mga paghihirap na nararanasan ng bata kapag siya ay bumangon mula sa isang posisyong nakaupo sa sahig. Sa sapat na malubhang anyo ng muscular dystrophy, ang bata ay may sintomas ng Govers: bumangon siya mula sa sahig, una sa lahat ay lumuhod siya, nakasandal sa kanyang mga kamay, at pagkatapos ay bumangon, sunud-sunod na itinutulak ang kanyang mga kamay mula sa kanyang mga buto, kasukasuan ng tuhod at hita (Larawan 21-9). Maaari mong matukoy ang kahinaan ng mga kalamnan ng sinturon sa balikat sa pamamagitan ng paghawak sa bata sa isang nakataas na posisyon sa pamamagitan ng mga kilikili. Karaniwan, sinusubukan niyang kumapit sa pamamagitan ng pagdiin ng kanyang mga braso sa kanyang katawan; na may muscular dystrophy, parang dumulas sa kamay ng examiner. Ang isang maysakit na bata ay madalas na hindi maaaring itaas ang kanyang mga braso sa itaas ng kanyang ulo. Sa mga huling yugto ng sakit, bubuo ang makabuluhang pagkasayang ng kalamnan. Karaniwan, sa edad na 12, ang isang bata ay hindi na makalakad. Ang mga pasyente sa 75% ng mga kaso ay namamatay bago ang edad na 20 taon. Karamihan sa kanila ay may cardiomyopathy, na sa ilang mga kaso ay sanhi biglaang kamatayan. Kung ang mana ay X-linked, at ang sakit ay nagsimula sa mas matandang pagkabata, ang pag-asa sa buhay ay nananatiling mahaba (Becker muscular dystrophy). Ang average na IQ para sa mga batang may Duchenne ay 80; 25% ng mga bata ay may mental retardation.
Sa differential diagnosis Dapat kasama sa Duchenne muscular dystrophy ang Werdnig-Hoffmann disease sa mas matatandang mga sanggol at mga sakit sa kalamnan tulad ng endocrine myopathies, carnitine deficiency, glycogen storage disease, at polymyositis. Minsan sa mga contracture ng calcaneal tendon at ang bata na naglalakad sa mga daliri ng paa, ang cerebral palsy ay maaaring ipagpalagay, ngunit sa muscular dystrophy ay walang mga katangian na palatandaan ng cerebral palsy spasticity at hyperreflexia.
Ang diagnosis ay batay sa pagpapasiya ng aktibidad ng serum enzyme, data ng electromyography at biopsy ng tissue ng kalamnan. Ang aktibidad ng mga enzyme, lalo na ang creatine phosphokinase, kahit na bago ang pag-unlad klinikal na sintomas madalas na lumampas sa pamantayan ng 10 beses kahit na sa mga sanggol. Sa electromyogram, una sa lahat, ang isang pagbawas sa tagal at isang pagbawas sa amplitude ng mga potensyal na motor ay ipinahayag. Ang mga pagbabago sa histological ay binubuo ng pagkabulok ng mga fibers ng kalamnan. Madalas silang nag-iiba sa laki at bahagyang pinapalitan ng taba at connective tissue. Ang laki ng kanilang nuclei ay nag-iiba din. Ang diagnosis ay maaaring maitatag sa kapanganakan sa pamamagitan ng pagtukoy sa aktibidad ng creatine phosphokinase. Ang mga pamamaraan para sa pagtukoy ng mga babaeng carrier ay hindi pa binuo, sa kabila ng katotohanan na 60-80% sa kanila ay nagpapakita ng bahagyang o katamtamang pagtaas sa antas nito. Ang mga palatandaang ito ay mas karaniwan sa pagkabata kaysa sa mga susunod na yugto ng buhay.
mabisang pamamaraan walang lunas. Ang pasyente ay dapat manatiling aktibo at makalakad hangga't maaari. Ito ay kinakailangan upang matiyak na ang bata ay umiiwas sa matinding pisikal na aktibidad, dahil maaari itong maging sanhi ng pagkalagot ng mga fibers ng kalamnan. Sa ilang mga kaso, ang pagpapahaba ng kirurhiko ng calcaneal tendon ay nagpapabuti sa kakayahang maglakad, ngunit matagal. pahinga sa kama pagkatapos ng orthopedic correction ay maaaring tumaas pananakit ng kasukasuan. Ang genetic counseling ay may mahalagang papel.
Congenital muscular dystrophy. Ang sakit ay minana sa isang autosomal recessive na paraan at nailalarawan sa pamamagitan ng hypotension ng kalamnan at kahinaan sa sanggol. Ito ay kasama sa pangkat ng mga kondisyon na tinukoy bilang "tamad na bata" (tingnan ang Talahanayan 21-1). Ang simula ng sakit ay tumutukoy sa intrauterine period. Minsan ang isang bagong panganak ay may binibigkas na pagkasayang ng mga kalamnan, ang kanilang mga contracture, limitadong magkasanib na kadaliang kumilos. Ang pagkakaiba sa sakit na Werdnig-Hoffmann ay mahirap. Ang mga fasciculations ng dila, na katangian ng huli, ay wala sa muscular dystrophy. Ang mga tendon reflexes ay nalulumbay, ngunit hindi ganap na nawala. Ang proseso ay nagsasangkot ng mga kalamnan na kasangkot sa paghinga, kabilang ang diaphragm. Sa matinding kaso, ang kamatayan ay nangyayari bago ang edad na 1 taon dahil sa respiratory failure; sa mas banayad na mga anyo, ang normal na posibilidad na mabuhay ay pinananatili sa loob ng mahabang panahon. Ang isang pagtaas sa aktibidad ng serum enzymes ay hindi nabanggit, bagaman ang mga dystrophic na pagbabago ay nangyayari sa mga kalamnan.
shoulder-facial form ng muscular dystrophy. Tama na ito banayad na anyo Ang muscular dystrophy ay minana sa isang autosomal dominant na paraan. Karaniwan itong nagsisimula sa edad na 10-20 taon at nailalarawan sa pamamagitan ng kahinaan at pagkasayang ng mga kalamnan ng mukha at sinturon sa balikat. Ang mukha ay ganap na amimic, ang pasyente ay hindi maaaring isara ang kanyang mga mata at gumawa ng isang sipol. Ang sakit ay umuunlad nang dahan-dahan at katugma sa normal na pag-asa sa buhay. Ang diagnosis ay batay sa mga klinikal na natuklasan at uri ng mana. Ang mga resulta ng isang biopsy ng kalamnan tissue ay nagpapahiwatig ng mga dystrophic na pagbabago dito. Maaaring manatiling normal o bahagyang tumaas ang mga antas ng serum creatine phosphokinase.
Pelvic form ng muscular dystrophy. Ang grupong ito ng mga heterogenous disorder ay nailalarawan sa pamamagitan ng isang mabagal na pag-unlad ng muscular dystrophy at minana sa isang autosomal recessive na paraan. Ang simula ng sakit ay tumutukoy sa mas matandang pagkabata, pagbibinata o pagtanda. Ang mga kalamnan ng pelvic girdle ay kadalasang apektado.
Ocular form ng myopathy. Ang mga dystrophic na pagbabago ay nangyayari pangunahin sa mga panlabas na kalamnan ng mata. Ang sakit ay nagsisimula sa pagkabata o pagbibinata. Kasama nito, umuunlad ang ptosis at limitasyon ng mga paggalaw. mga eyeballs. Minsan ang kahinaan ay umaabot sa mga kalamnan ng mukha at leeg. Ang sakit ay dapat na naiiba sa myasthenia gravis at paralysis cranial nerves na may mga tumor sa tangkay ng utak.
Ang progresibong ophthalmoplegia, simula sa pagkabata o pagbibinata, ay nauugnay sa atypical retinitis pigmentosa at heart block (Kearns-Sayers syndrome). Ito ay kadalasang nauugnay sa progresibong ataxia, naantalang paglaki at pagdadalaga. Sa ilalim ng sarcolemma ng mga kalamnan, ang malalaking akumulasyon ng hindi tipikal na mitochondria ay tinutukoy. Ang genetic na katangian ng prosesong ito ay hindi naitatag. Maaari mong kontrolin ang posibilidad ng biglaang pagkamatay mula sa isang paglabag sa cardiac conduction sa isang pacemaker.
Myotonic dystrophy. Sa kabila ng katotohanan na ang myotonic dystrophy ay nagsisimula na parang nasa isang may sapat na gulang, ang simula nito ay lalong naitala sa mga sanggol at mga bata sa ibang pagkakataon. Ito ay minana sa isang autosomal dominant na paraan. Ang simula nito sa pagkabata ay nagpapahiwatig na ang ina ay naghihirap mula sa myotonia. Alinsunod dito, ang mga kadahilanan ng intrauterine ay maaaring makaimpluwensya sa kalubhaan ng sakit sa isang bata. Nasa oras ng kapanganakan, maaari niyang matukoy muscular hypotension wala siyang kakayahang sumipsip. Ang lag ng pisikal at mental na pag-unlad ay karaniwang nakikita sa ibang pagkakataon. Sa maagang pagkabata, ang kahinaan ng kalamnan at pagkasayang ay pangunahing kumalat sa mga kalamnan ng mukha, panga at temporal. Karaniwang napapansin ang bilateral ptosis. Kasama sa mga diagnostic na makabuluhang pamamaraan ang muscle percussion, electromyography; tipikal para sa mga pasyenteng ito ay ang kawalan ng kakayahan na alisin ang kamay na nakakuyom sa isang kamao (tingnan ang Congenital myotonia). Ang kahinaan at pagkasayang ng mga kalamnan ng mga limbs at pelvic girdle (karaniwan ay distal na grupo) ay nakikita sa mas matandang pagkabata o pagbibinata. Sa mga matatanda, ang sakit na ito ay sinamahan ng mga katarata, pagkakalbo, testicular atrophy.
Ang diagnosis ay batay sa pagkakakilanlan ng mga palatandaan ng myotonia, ang katangian ng pamamahagi ng kahinaan ng kalamnan, pamana ayon sa nangingibabaw na uri, at mga dystrophic na pagbabago sa mga kalamnan. Sa pagkabata, ang kurso ng sakit ay maaaring hindi kanais-nais, madalas na sinamahan ng mental retardation. Sa pagbibinata, nauuna ang kahinaan ng kalamnan. Sa mga functional disorder, ang paggamot na may novocaine at quinidine ay ipinahiwatig.

