Albrightov syndróm - zriedkavé genetické ochorenie s neznámym pôvodom. Dedí sa a vyznačuje sa najmä poškodením kostrový systém. Najčastejšie sa vyskytuje u žien. Ale sú chvíle, keď môžu ochorieť aj chlapci. Väčšina pacientov s týmto syndrómom trpí telesnou a mentálnou retardáciou.
Choroba bola pomenovaná po vedcovi, ktorý ju objavil. Kostrový systém sa začína rozpadať pod vplyvom metabolizmu vápnika a fosforu. Prištítna žľaza vylučuje parathormón, u osoby s Albrightovým syndrómom je narušená odolnosť tkaniva voči parathormónu.
Symptómy syndrómu Butler Albright
Existujú tri hlavné funkcie choroby. Ak sú prítomní čo i len dvaja z troch, možno takúto diagnózu stanoviť.
- Veľmi skorá puberta v dôsledku vysoko zvýšených hormonálnych hladín. Dievčatá môžu začať menštruovať v prvých mesiacoch po narodení. Chlapci s Albrightovým syndrómom veľmi zriedkavo zažijú skorú pubertu. Zmeňte sa hormonálne pozadie sa vyskytuje v dôsledku nesprávneho fungovania nadobličiek, hypofýzy a štítnej žľazy. Tento príznak sa môže objaviť v úplnej alebo neúplnej forme. V plnej forme začína menštruácia veľmi skoro a je zvyčajne silná a bolestivá. V dojčenskom veku sa tento syndróm prejavuje aj sekundárnymi pohlavnými znakmi, napríklad opuchom mliečnych žliaz. Ak je prejav neúplný, menštruácia sa nevyskytuje. Veľmi často sa pri Albrightovom syndróme u dievčat vyvinú cysty na vaječníkoch. U chlapcov sa počas skorej puberty pozoruje zväčšenie penisu a rast pubického ochlpenia.
- Fibrózna osteodysplázia. Toto ochorenie naznačuje porušenie kostného tkaniva, čo vedie k zakriveniu a deformácii kostí. Charakterizovaná asymetriou. Najčastejšími znakmi sú krívanie a zakrivenie chrbtice. Najčastejšie sú zmenami postihnuté dlhé kosti. Rast kostry sa spomaľuje, je to badateľné aj u detí. U mnohých obetí McCuneovho syndrómu dochádza k nerovnomernému rastu kostí lebky, čo vedie k vonkajšej deformácii. Keď sa tvár zmení, môžu sa objaviť vypuklé oči. Po dosiahnutí veku puberty kostné zmeny spomaliť a výskyt zlomenín sa zníži.
- Zmeny na koži. Prejavuje sa zvýšenou pigmentáciou. Koža na trupe a stehnách je pokrytá žltohnedými škvrnami so zubatými okrajmi. Pigmentáciu možno vidieť na spodnej časti chrbta, hornej časti chrbta, zadku a hrudníka. Škvrny sa objavia ihneď po narodení dieťaťa.

Liečba Albrightovej (McCune) syndrómu
 Pacienti trpiaci McCuneovým syndrómom, musí byť neustále pod dohľadom špecializovaných odborníkov (gynekológ, endokrinológ, traumatológ, oftalmológ).
Pacienti trpiaci McCuneovým syndrómom, musí byť neustále pod dohľadom špecializovaných odborníkov (gynekológ, endokrinológ, traumatológ, oftalmológ).
Nevyhnutné komplexná liečba. Keďže choroba je charakterizovaná genetická mutácia, potom je potrebné v prvom rade zbaviť sa príznakov jej prejavu.
Povinné zobrazené hormonálna liečba na udržanie endokrinného systému.
Aby sa predišlo týmto zmenám počas choroby, je potrebné nepremeškať okamih, keď sa kosti tváre začnú deformovať.
Niekedy sa to vyžaduje chirurgická intervencia . Predpisuje sa, keď v dôsledku zmien v štruktúre lebky existuje riziko straty sluchu alebo zraku.
Počas bolesti sa používajú špeciálne lieky proti bolesti obsahujúce malé množstvá bisfosfonátov.
Je veľmi dôležité vykonať preventívne opatrenia na posilnenie svalov.
Pre každého pacienta, ktorý trpí Albrightovou chorobou, lekár vyberá vlastnú individuálnu liečbu, ktorú je potrebné prísne dodržiavať.
Musí byť neustále monitorovaný množstvo vápnika v tele. Pri začatí liečby sa takéto vyšetrenia vykonávajú raz týždenne, potom sa ich frekvencia zníži na raz za mesiac, kým sa nedokončí hlavná liečba.
Počas liečby je veľmi dôležité dodržiavať diétu, pri ktorej je množstvo potravín s obsahom fosforu znížené na minimum.
Treba poznamenať, že žena trpiaca týmto syndrómom Pri rozhodovaní o otehotnení by ste sa určite mali poradiť s dobrým genetikom.
Albrightova choroba (McCune) sa vyskytuje u jedného človeka z 1 000 000. Špecifická liečba tohto syndrómu ešte nebola vynájdená, no netreba strácať nádej. Medicína nestojí na mieste a lekári pomôžu prekonať nepríjemné chvíle a budú mať pacienta neustále pod dohľadom.
Gény sú zdrojom všetkých informácií o stavbe a fungovaní tela. Od ich správneho fungovania závisí doslova všetko: tvorba orgánov a tkanív, obnova poškodených štruktúr a tvorba nových, ako aj metabolizmus. Všetky chemické premeny v tele sú zaznamenané v génoch. Metabolizmus prebieha pomocou špeciálnych urýchľovačov – enzýmov. Ich nesprávna štruktúra vedie k mnohým problémom. Medzi podobné dedičné patológie patrí Albrightova choroba.
Výmena vápnika a fosforu v tele
Metabolizmus je najdôležitejšou súčasťou života ľudského tela. Každú sekundu len v tkanivách a orgánoch chemické prvky premeniť sa na iných. Ich obsah je spravidla prísne regulovaný. To platí najmä pre elektrolyty – sodík, draslík, vápnik a fosfor. Práve vďaka ich špecifickým vzťahom je možná práca srdca a kostrového svalstva a prenos nervového elektrického signálu.
Vápnik sa nachádza vo všetkých orgánoch a tkanivách a hrá niekoľko dôležitých úloh:
- je stavebným materiálom pre kosti;
Vápnik je hlavným stavebným prvkom kostného tkaniva
- zabezpečuje fungovanie srdca a kostrových svalov;
Vápnik je jedným z účastníkov svalovej kontrakcie
- podieľa sa na zrážaní krvi a metabolizme;
- je sprostredkovateľom pôsobenia hormónov – špeciálnych chemických signálov, ktoré regulujú fungovanie organizmu.
Fosfor sa nachádza vo veľkých množstvách v kostnom tkanive a je nevyhnutný pre normálnu funkciu mozgu. Vápnik a fosfor sú v krvi prítomné v presne definovanom pomere. Prebytok týchto chemických zlúčenín sa vylučuje obličkami močom.
Parathormón je hlavným regulátorom hladín vápnika a fosforu v krvi
Pohoda tela priamo závisí od konštantného množstva vápnika a fosforu. Proces ich výmeny je riadený štítnou žľazou a prištítnymi telieskami:
- Prvý produkuje hormón kalcitonín, vďaka ktorému sa vápnik prenáša z krvi do kostného tkaniva.
- Posledne menované pomocou parathormónu eliminujú nedostatok vápnika v krvi vymývaním z kostí.
Okrem toho je výmena vápnika a fosforu regulovaná obličkami, čím sa prebytok týchto chemických zlúčenín odstraňuje močom. Významnú úlohu v tom zohráva vitamín D.
Albrightov syndróm je dedičné ochorenie spôsobené poruchou metabolizmu vápnika a fosforu v tele. Vedie k rôznym zmenám kostí, obličiek, tráviaceho traktu a duševným poruchám.
Synonymá pre Albrightovu chorobu: pseudohypoparatyreóza, dedičná osteodystrofia.
O dedičnej osteodystrofii prvýkrát uvažoval vedec Fuller Albright v roku 1942. Choroba je niekoľkonásobne častejšia u žien. IN lekárska literatúra Bolo popísaných len asi 300 prípadov tejto zriedkavej patológie.
Klasifikácia
Pseudohypoparatyreóza sa delí na tri hlavné typy:

Príčiny a faktory vývoja
Pseudohypoparatyreóza je založená na dedičnej rezistencii telesných tkanív na vplyv parathormónu. Ten za normálnych podmienok pôsobí prostredníctvom špeciálneho sprostredkovateľa – cyklického adenozínmonofosfátu (cAMP). Je to on, kto dáva pokyn bunke, aby poslúchla regulačný signál parathormónu. Pri Albrightovej chorobe sa štruktúra mediátora mení na genetickej úrovni.
Albrightova choroba sa prenáša na ďalšie generácie podľa zaujímavého vzoru, ktorý sa nazýva X-viazaná dominantná dedičnosť: patológia prechádza z matky na dcéry a synov v 50% prípadov. S rovnakou pravdepodobnosťou dedia defektný gén po otcovi iba dcéry. Existujú rodinné prípady ochorenia.
Gén Albrightovho syndrómu sa nachádza na pohlavnom chromozóme
Bez regulačného pôsobenia parathormónu sa metabolizmus vápnika výrazne posúva smerom k využitiu do kostného tkaniva. V tomto prípade je jeho nedostatok v krvi, čo vedie k nasledujúcim javom:
- svalové kŕče;
- narušenie tvorby elektrických impulzov v myokarde a zmeny srdcového rytmu;
- zvýšená produkcia parathormónu a zväčšenie prištítnych teliesok.
Nadbytok parathormónu zase vedie k zvýšeniu hladiny fosforu v krvi.
Defektný enzým pri Albrightovej chorobe narúša aktivitu nielen parathormónu. Patológia zahŕňa hormóny štítnej žľazy a pohlavných žliaz vo svojom začarovanom kruhu. V niektorých prípadoch nastáva skorá puberta – puberta – v dôsledku vysokej aktivity pohlavných hormónov.
Štítna žľaza a prištítne telieska sú hlavnými regulátormi metabolizmu vápnika a fosforu v tele.
Albrightova choroba - video
Príznaky Albrightovej choroby
Dedičná choroba najčastejšie postihuje celé telo, čo vedie k viacerým zmenám. Príznaky patológie sú v tomto prípade mimoriadne podobné príznakom hypoparatyreózy - nedostatku produkcie parathormónu v prištítnych telieskach.
Bežným príznakom pseudohypoparatyreózy sú svalové kŕče. Ich závažnosť priamo závisí od hladiny vápnika v krvi: od zášklbov jednotlivých vlákien až po celkový spazmus celého kostrového svalstva – opistotonus.
Príznaky pseudohypoparatyreózy - tabuľka
| Lokalizácia zmien | Kostra | Kostrové svaly | Srdcový sval | Obličky | Mozog a zmyslové orgány | Koža, tukové a spojivové tkanivo |
| Príznaky choroby |
|
|
|
|
| Tvorba subkutánnych akumulácií kryštálov vápnika |
Diagnostické metódy
Na rozpoznanie Albrightovej choroby je potrebná spoločná práca niekoľkých odborníkov: urológ, neurológ, psychiater, endokrinológ, genetik. Na stanovenie správnej diagnózy sa používajú tieto metódy:
- objektívne vyšetrenie - umožňuje identifikovať nízky vzrast, anomálie v štruktúre rúk a zubov;
Albrightova choroba spôsobuje skrátenie prstov
- neurologické vyšetrenie – pomáha odhaliť príznaky zvýšenej kŕčovej pripravenosti v podobe zášklbov jednotlivých svalov tváre, trupu a končatín;
- vyšetrenie u psychiatra - umožňuje zistiť mentálnu retardáciu, poruchy intelektu a pamäti;
- biochemický krvný test – umožňuje zistiť nízke hladiny vápnika, vysoké hladiny fosforu a normálne alebo zvýšené hladiny parathormónu;
Vápnik a fosfor sú jedným z hlavných prvkov tekutej časti krvi
- biochemický test moču – zisťuje nízke hladiny vápnika a fosforu;
- test so zavedením parathormónu - umožňuje rozlíšiť pseudohypoparatyreózu od skutočného nedostatku parathormónu. Pri Albrightovej chorobe nespôsobuje zavedenie parathormónu do krvi zvýšenie množstva fosforu v moči;
- Röntgenové meranie hustoty kostí (denzitometria) – diagnostikuje anomálie v štruktúre ruky, oblasti mäknutia a rednutia kostného tkaniva, ako aj akumuláciu vápnika vo svaloch, koži a podkoží;
Pri Albrightovej chorobe sa mení hustota kostnej hmoty (vľavo - normálna, vpravo - patológia)
- počítačové (alebo magnetické rezonančné) zobrazovanie mozgu - umožňuje identifikovať ložiská akumulácie vápnika v jednotlivých štruktúrach, napríklad bazálnych gangliách, ktoré sú zodpovedné za koordináciu pohybov;
Vápnik sa často ukladá v oblasti bazálnych ganglií
- Ultrazvukové vyšetrenie obličiek - pomáha určiť prítomnosť kameňov, ich počet a veľkosť;
- Ultrazvuk prištítnych teliesok môže odhaliť zvýšenie veľkosti orgánu, ako aj prítomnosť cýst.
Odlišná diagnóza vykonávané s nasledujúcimi chorobami:

Liečebné metódy
Pseudohypoparatyreóza vyžaduje celoživotné sledovanie endokrinológom. Pri opakovaných ťažkých záchvatoch kŕčov je nutná hospitalizácia na špecializovanom oddelení nemocnice.
Medikamentózna terapia
Liečba liekmi je hlavnou zložkou terapie pseudohypoparatyreózy. Hlavnými cieľmi sú:
- normalizácia hladín vápnika a fosforu v krvi;
- obnovenie normálnej štruktúry kostí;
- prevencia záchvatov;
- prevencia tvorby obličkových kameňov.
Na normalizáciu hladín vápnika sa používajú lieky Chlorid vápenatý a Glukonát vápenatý. Najčastejšie sa tieto lieky používajú vo forme tabliet. Na odstránenie svalových kŕčov sa však praktizuje intravenózne podanie farmakologické látky.
Glukonát vápenatý je liek používaný na liečbu Albrightovej choroby
Doplnky vitamínu D pomáhajú normalizovať hladiny vápnika a fosforu v krvi:
- Oksidevit;
- kalcitrín.
Vitamín D tiež pomáha posilňovať kostné tkanivo a zuby.
Pri Albrightovej chorobe je predpísaný vápnik D3
Podávanie parathormónu pri pseudohypoparatyreóze je neúčinné, pretože tkanivá a orgány sú necitlivé na jeho vplyv.
Korekcia stravy
Dôležitou zložkou liečby pseudohypoparatyreózy je úprava stravy. Zároveň je životne dôležitý dostatočný kalorický príjem a frakčné jedlá. Množstvo tekutín, ktoré denne vypijete, by malo byť aspoň 1,5 – 2 litre.
- fazuľa;
- zelený hrach;
- šošovica;
- mandle;
- žerucha;
- zeler;
- karfiol;
- kôpor;
- bazalka;
- mlieko;
- jogurt;
- kyslá smotana.
Potraviny s vysokým obsahom vápnika - fotogaléria
 Fazuľa má vysoký obsah vápnika
Fazuľa má vysoký obsah vápnika  Sója je šampiónom medzi strukovinami v obsahu vápnika (277 mg na 100 g)
Sója je šampiónom medzi strukovinami v obsahu vápnika (277 mg na 100 g)  Žerucha je zelená s vysokým obsahom vápnika (81 mg na 100 g)
Žerucha je zelená s vysokým obsahom vápnika (81 mg na 100 g)  Zeler sa používa na prípravu zeleninových šalátov
Zeler sa používa na prípravu zeleninových šalátov  Bazalka sa odporúča používať ako korenie
Bazalka sa odporúča používať ako korenie  Fermentované mliečne výrobky obsahujú veľa vápnika
Fermentované mliečne výrobky obsahujú veľa vápnika
Potraviny s vysokým obsahom fosforu, ktorých konzumácia by mala byť obmedzená:
- sezamové semienka;
- morské ryby;
- žĺtok;
- mandle;
- pistácie;
- Píniové oriešky;
- arašidy;
- Orech.
Produkty s vysokým obsahom fosforu - fotogaléria
 Morské ryby – zdroj veľká kvantita fosfor
Morské ryby – zdroj veľká kvantita fosfor  Konzumácia vaječných žĺtkov v prípade patológie by mala byť obmedzená.V prípade Albrightovej choroby je lepšie zdržať sa cíceru.
Konzumácia vaječných žĺtkov v prípade patológie by mala byť obmedzená.V prípade Albrightovej choroby je lepšie zdržať sa cíceru.  Vlašské orechy líšiť vysoký stupeň fosfor
Vlašské orechy líšiť vysoký stupeň fosfor
Chirurgia
Neexistuje žiadna chirurgická liečba pseudohypoparatyreózy. V niektorých podmienkach sprevádzajúcich priebeh ochorenia je potrebná pomoc chirurga:

Fyzioterapia
Fyzioterapeutické opatrenia sa používajú pri liečbe niektorých stavov sprevádzajúcich priebeh Albrightovej choroby:
- so zápalom pohárov a panvy obličiek (pyelonefritída) na pozadí prítomnosti kameňov;
- so zápalom ložísk akumulácie vápnika vo svaloch a podkožnom tuku;
- na hojenie zlomenín.
Na odstránenie týchto negatívnych javov sa používajú tieto metódy:

Komplikácie a prognóza
Prognóza terapie s včasným rozpoznaním Albrightovej choroby je relatívne priaznivá, ale nie je možné úplne vyliečiť dedičný defekt enzýmu. Normalizácia obsahu vápnika a fosforu v tele umožní pacientom zapojiť sa do ľahkej práce, s výnimkou nervové napätie a práca so strojmi a mechanizmami.
Pri opakovaných záchvatoch svalových kŕčov, ktoré sa ťažko upravujú liekmi, rozhoduje lekárska komisia o pridelení skupiny postihnutia. V závažných prípadoch pseudohypoparatyreózy sa môžu vyvinúť nasledujúce komplikácie:

Prevencia
Hlavná preventívne opatrenie pre pseudohypoparatyreózu je lekárske genetické poradenstvo. Genetik bude študovať rodokmeň manželov a určí pravdepodobnosť patológie u potomkov.
Pseudohypoparatyreóza je závažné dedičné ochorenie zahŕňajúce patologický proces všetky tkanivá a orgány. Včasná konzultácia s lekárom a dodržiavanie liečebných odporúčaní umožní pacientovi udržať si schopnosť pracovať a kvalitu života.
Prvýkrát v ruská literatúra tento syndróm opísal V. R. Braitsev v roku 1922, pričom ho definoval ako "vláknité nádory" O 15 rokov neskôr endokrinológ Fuller Albright sformuloval znaky tohto systémového ochorenia ako „syndróm charakterizovaný diseminovanou osteitis fibrosa, pigmentovými poliami a endokrinnými poruchami s predčasnou pubertou u dievčat“.
Inými slovami, McCune-Albright-Braitsevov syndróm je systémové ochorenie, charakterizované predčasným sexuálnym vývojom a kostnou patológiou. Toto ochorenie sa vyskytuje prevažne u žien a je to vrodená dedičná patológia. Donedávna sa verilo, že ide o výlučne ženské ochorenie, no potom boli prípady ochorenia zaznamenané aj u chlapcov. Za príčinu syndrómu sa považuje mutácia v špecifickom géne.
Známky McCune-Albright-Braitsevovho syndrómu
- Tmavé škvrny. Všetci pacienti majú špeciálnu pigmentáciu nepravidelného tvaru, farby kávy s mliekom. Škvrny sa zvyčajne nachádzajú na chrbte, hrudníku, stehnách, zadku a spodnej časti chrbta. Toto je druh markera choroby; pigmentácia je prítomná od narodenia alebo sa objavuje, keď dieťa rastie.
- Patológia kostí. Zvyčajne sú postihnuté dlhé kosti (napríklad stehenná kosť). Prítomnosť dutín v kostiach vedie k ich deformácii, zlomeninám a ohýbaniu končatín. Typickou deformitou bedra je typ „pastiersky krívačky“. Možné poškodenie kostí lebky. V tomto prípade dochádza k asymetrii tváre a jednostrannému exoftalmu (vypuklé oči). Poškodenie kostí a počet zlomenín klesá po puberte.
- Predčasný sexuálny vývoj. Môže začať vo veku 6-9 mesiacov až do 7 rokov. Najčastejšie sa prvé známky puberty objavujú u dieťaťa vo veku 3 rokov. Môže sa prejaviť v úplných a neúplných formách. Plná forma u dievčat sa vyznačuje menarché (prvá menštruácia) a zväčšením mliečnych žliaz. Dlhodobé a silné krvácanie, cysty sa často nachádzajú na vaječníkoch. V neúplnej forme sa menarché nevyskytuje.
Predčasná puberta u chlapcov je oveľa menej častá a je sprevádzaná symetrickým zväčšením semenníkov, potom rastom penisu a ochlpenia, rovnako ako pri normálnom sexuálnom vývoji.
- Akcelerácia kostného veku a zrýchlené uzatváranie rastových platničiek.
- Neurologické a mentálne poruchy(mentálna retardácia, kŕče, atrofia optický nerv, strata sluchu).
- V literatúre sú informácie o ďalších endokrinných poruchách pri tomto ochorení: akromegália, hyperparatyreóza (zvýšená funkcia prištítnych teliesok), hypertyreóza (zvýšená funkcia štítnej žľazy) a prítomnosť uzlín v štítna žľaza, hyperkortizolizmus (nadbytok glukokortikoidných hormónov nadobličiek), krivica odolná voči vitamínu D.
Diagnóza syndrómu McCune-Albright-Braitsev
- Prítomnosť charakteristických znakov.
- Genetická konzultácia.
- Röntgenové vyšetrenie.
- Pohlavné hormóny a gonadotropíny, hormóny štítnej žľazy, prolaktín, iné hypofyzárne tropické hormóny (podľa indikácií).
- Ultrazvuk panvových orgánov pre dievčatá a miešok pre chlapcov.
- Konzultácia s endokrinológom a detským gynekológom u dievčat alebo s detským urológom u chlapcov.
- Vyšetrenie iných orgánov endokrinného systému (ako je uvedené).
Liečba syndrómu McCune-Albright-Braitsev
Pacienti s touto patológiou potrebujú dohľad nad gynekológom-endokrinológom, endokrinológom, lekárom rádiologická diagnostika, oftalmológ, ortopéd-traumatológ. Okrem toho je potrebné periodicky určovať úroveň tropického a gonadotropné hormóny. Špecifická liečba tento syndróm nebol vyvinutý. Najčastejšie lekári volia pozorovaciu taktiku. U dievčat je terapia indikovaná v prípade dlhotrvajúceho zvýšenia hladín estrogénu, sprevádzaného častým a závažným krvácaním. Podľa indikácií sa vykonáva korekcia kostných deformácií. Keď sa objavia príznaky poškodenia iných Endokrinné žľazy liečbu vykonávajú endokrinológovia.
Albrightova dedičná osteodystrofia (synonymá: pseudohypoparatyreóza typu 1a, Albrightova choroba, Albrightov syndróm) je zriedkavé ochorenie kostrového systému spôsobené mutáciou génu lokalizovaného na 20. chromozóme. Patológia je charakterizovaná porušením metabolizmu vápnika a fosforu, čo ovplyvňuje oneskorenie fyzického a niekedy aj duševného vývoja. Spôsob dedičnosti Albrightovho syndrómu je stále predmetom diskusií.
Pre informáciu: chorobu ako prvý opísal v roku 1942 americký endokrinológ F. Albright. V súčasnosti je výskyt patológie 79 prípadov na 10 miliónov ľudí. Ženy ochorejú dvakrát častejšie ako muži.
Pseudohypoparatyreóza: popis a symptómy
Ochorenie napodobňuje hypoparatyreózu – nedostatok parathormónu (parathormónu). Mechanizmus výskytu sa však líši v tom, že nedochádza k nedostatku hormónu, ale genetický defekt v komplexe bunkových receptorov v tkanivách obličiek a kostry blokuje jeho pôsobenie. Metabolické procesy, ktoré sú normálne regulované parathormónom prostredníctvom syntézy cAMP, sú nemožné. Prištítne telieska môžu byť kompenzačne zväčšené.

V závislosti od typu ochorenia (a teraz sú štyri klinické formy: typ 1 a 2, prvý je tiež rozdelený na podtypy A, B a C), sú možné rôzne možnosti blokovania účinku parathormónu, ako aj zhoršená rezistencia na iné hormóny: glukagón, TSH, GnRH, ADH. V dôsledku toho sa vyvíjajú javy hypotyreózy, amenorea a koncentračná funkcia obličiek je narušená.
Zvyšuje sa množstvo fosforu v krvi, klesá hladina aktívnej formy vitamínu D3 a vápnika.
Vzhľad pacientov s Albrightovým syndrómom
Ochorenie vedie k viacerým abnormalitám vývoja kostry, ktoré sa nachádzajú v detstva, niekedy po roku, ale zvyčajne v 5-10 rokoch a aj neskôr. Môžete mať podozrenie na Albrightovu chorobu u dieťaťa na základe nasledujúcich príznakov:
- tvár v tvare mesiaca;
- krídlové záhyby na krku;
- nízky rast v dôsledku skrátených nôh;
- obezita;
- skrátenie prstov na rukách a nohách (brachydaktýlia);
- aplázia (nedostatočný vývoj) zubov;
- časté zlomeniny kostí;
- predčasný sexuálny vývoj:
- viaceré podkožné oblasti kalcifikácie.

Dôležité! Ktorýkoľvek z týchto príznakov vyžaduje kontaktovanie endokrinológa, predpisovanie diagnostické opatrenia identifikovať príčiny tohto stavu a zvoliť adekvátnu liečbu. Od 90. rokov minulého storočia sa diagnostika vykonáva na základe metód molekulárnej genetiky.
Včasný kontakt s endokrinológom a genetikom odďaľuje správnu diagnózu a možnosť liečby.
Zmeny v kostiach a mäkkých tkanivách
Zmeny podobné prejavom hypoparatyreózy sa nachádzajú v kostnom tkanive:
- difúzna osteoporóza;
- cysty (hnedé alebo obrovské bunkové nádory).
Vápnik sa uvoľňuje z kostí a vytvára kalcifikácie v mäkkých tkanivách:
- v podkožnom tkanive;
- svaly vrátane myokardu;
- obličky;
- v rohovke a spojovke očí;
- steny hlavných tepien.
Na koži sa objavujú oblasti hyperpigmentácie.
Iné znaky
Porušenie metabolizmu vápnika vedie k záchvatom a zvracaniu. V niektorých prípadoch sa pozoruje hematúria v dôsledku tvorby oxalátových kryštálov v močovom trakte.

Mentálna retardácia, cukrovka, šedý zákal môže byť aj prejavom Albrightovej choroby.
Diagnostika
Diagnóza je založená na analýze klinických prejavov, röntgenovom vyšetrení kostí a mäkkých tkanív a detekcii pseudohypoparatyreózy pomocou biochemických testov.
Krvné testy ukazujú hypokalciémiu a hyperfosfatémiu, hladiny alkalickej fosfatázy v sére sú normálne alebo zvýšené, rovnako ako PTH.
Laboratórnym dôkazom pseudohypoparatyreózy je absencia zvýšenia hladiny nefrogénneho cAMP a fosfátu v moči ako odpoveď na podanie parathormónu.
Možnosti liečby
Na rozdiel od skutočnej hypoparatyreózy nemá pri Albrightovej chorobe podávanie parathormónu žiadny účinok.
Cieľom liečby je vrátiť hladinu vápnika a vitamínu D v krvi do normálu. K tomu predpíšte lieky obsahujúce tieto látky: dihydrotachysterol, oxydevit, kalcitrín.
Catad_tema Pediatria - články
Pseudohypoparatyreóza (Albrightova dedičná osteodystrofia): ťažkosti diferenciálneho diagnostického hľadania. Klinické pozorovanie
E.V. Tozliyan, detský endokrinológ, genetik, Ph.D., I.V. Shulyakova, neurológ, Ph.D.,
izolovaný štrukturálne členenie"Výskumný klinický ústav pediatrie" Štátna rozpočtová vzdelávacia inštitúcia vyššieho odborného vzdelávania "Ruská národná výskumná lekárska univerzita pomenovaná po N.I. Pirogov“ z Ministerstva zdravotníctva Ruskej federácie, Moskva
Kľúčové slová:
deti, pseudohypoparatyreóza, Albrightova dedičná osteodystrofia, obezita, hypokalciémia, diagnostika, rezistencia na parathormón.
Kľúčové slová: deti, pseudohypoparatyreóza, Albrightova dedičná osteodystrofia, obezita, hypokalciémia, diagnostika, rezistencia na parathormón.
Pseudohypoparatyreóza (grécky pseudes - nepravda + hypoparatyreóza; synonymum: Albrightova dedičná osteodystrofia, syndróm Javanese chicken) je zriedkavé dedičné ochorenie kostrového systému, simulujúce hypoparatyreózu a charakterizované poruchou metabolizmu vápnika a fosforu; často sprevádzané oneskoreným duševným a fyzickým vývojom. Toto ochorenie prvýkrát opísal americký endokrinológ Albright F. v roku 1942. Prevalencia ochorenia je 7,9 na 1 milión ľudí.
GENETICKÉ ÚDAJE
Pseudohypoparatyreóza (PHP) je geneticky heterogénne ochorenie. Údaje o type dedičného prenosu sú protichodné: dominantné aj autozomálne dominantné, autozomálne recesívne typy. Vo väčšine prípadov je rozvoj Albrightovej dedičnej osteodystrofie spojený s mutáciami v lokusu 20q13 génu GNAS1 lokalizovaného na chromozóme 20 (Patten et al., 1990), ktorý kóduje Gs-alfa proteín spojený s receptorom parathormónu (PTH). . Podobný fenotyp bol zistený aj u pacientov s intersticiálnou deléciou dlhého ramena chromozómu 2 v lokuse 2q37.
PATOGENÉZA
Patogenéza pseudohypoparatyreózy je založená na geneticky podmienenej rezistencii obličiek a skeletu na pôsobenie parathormónu v dôsledku defektu komplexu „špecifický cytoreceptor – parathormón – adenylátcykláza“, ktorý narúša tvorbu cyklických 3“- 5"-adenozínmonofosfát (cAMP) v obličkách, ktorý je intracelulárnym mediátorom účinku parathormónu na metabolické procesy. Pseudohypoparatyreóza je geneticky podmienená heterogénne ochorenie. U niektorých pacientov je defektný samotný cytoreceptor, ktorý viaže parathormón (pseudohypoparatyreóza typu 1A); iní majú defekt v proteíne viažucom nukleotidy lokalizovaného v lipidovej dvojvrstve bunková membrána a funkčná väzba receptora na adenylátcyklázu (pseudohypoparatyreóza typu 1B). Niektorí pacienti majú enzymatický deficit samotnej adenylátcyklázy (pseudohypoparatyreóza typu 2). Nedostatok cAMP vyplývajúci z týchto defektov vedie k narušeniu syntézy špecifických proteínov, ktoré určujú biologický účinok parathormónu. Tým sa stráca citlivosť cieľových orgánov na parathormón.
KLINICKÉ CHARAKTERISTIKY
V súčasnosti existujú 4 klinické formy patológie: typy 1A, 1B, 1C a 2. Znalosť ich klinických a biochemických znakov a údajov genetický výskum umožňuje diferenciálnu diagnostiku v rámci samotnej nozologickej formy.
Bežnými znakmi, ktoré umožňujú podozrenie na chorobu, sú disproporcionalita vo fyzickom vývoji, nízky vzrast (až do trpasličieho vzrastu) v dôsledku skrátenia dolných končatín (foto 1), brachydaktýlia (foto 2) a okrúhly „v tvare mesiaca“ tvár (foto 3). Niekedy sa pozorujú exostózy a dentálna aplázia.
Fotografia 1.
Vzhľad dieťa s Albrightovou osteodystrofiou
(vlastnosti fenotypu, nízky vzrast v dôsledku skrátenia dolných končatín)

Fotografia 2.
Vlastnosti kostrového systému pacienta
s Albrightovou osteodystrofiou
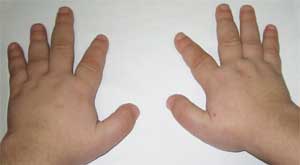
(brachydaktýlia - skrátenie prstov)
Fotka 3.
Vlastnosti fenotypu dieťaťa
s Albrightovou osteodystrofiou
(okrúhla tvár v tvare mesiaca)
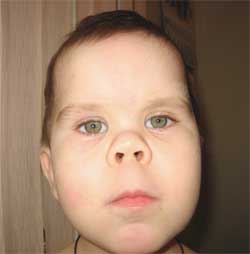
Patognomickým znakom je prudké skrátenie I, III a V metakarpálov a metatarzálnych kostí(najmä III a IV), v dôsledku čoho sú druhé prsty na rukách a nohách dlhšie ako ostatné a keď je ruka zovretá v päsť, v oblasti IV a V nie sú žiadne vydutiny. metakarpofalangeálne kĺby - takzvaný brachymetafalangizmus. Zisťujú sa aj krátke široké falangy, zhrubnutie lebečnej klenby a demineralizácia kostí (osteoporóza) a obezita.
Mentálna retardácia (zvyčajne stredne závažná) sa nachádza približne u 20 % pacientov. Podľa niektorých autorov sa mentálna retardácia vyskytuje v 70 % prípadov v prítomnosti hypokalcémie a v 30 % prípadov pri normokalciémii. Duševné procesy u pacientov sú spomalené. V neurologickom stave sa často zaznamenáva motorická nemotornosť a neurotické reakcie: strach, úzkosť, nepokoj, zlý sen, zvýšené reflexy, kŕče, ktoré majú tetanický charakter a sú spôsobené hypokalciémiou, niekedy kŕčovité záchvaty. Boli opísané aj myopatické symptómy: svalová únava, svalová slabosť. Často sa pozorujú extrapyramídové poruchy: choreiformná hyperkinéza, atetóza, tvárový hemispazmus, parkinsonizmus; v niektorých prípadoch sa vyskytujú epileptické paroxyzmy, cerebelárne symptómy: ataxia, strata koordinácie.
Často sa zistí kalcifikácia mäkkých tkanív a podkožné kalcifikácie (hrudník, brucho, šľachy päty), ktorých histologické vyšetrenie ukáže osteóm kože(Izraeli et al., 1992), mozog (bazálne gangliá). Je dôležité poznamenať, že kalcifikácie môžu byť prítomné už pri narodení. V dôsledku hypokalcémie zvyčajne vzniká šedý zákal a vznikajú defekty zubnej skloviny.
PSEUDOHYPOPARATHRÓZA TYPU 1A
má autozomálne dominantný vzor dedičnosti. Gén pre pseudohypoparatyreózu typu 1A – GNAS1 – je lokalizovaný na dlhom ramene chromozómu 20, v lokuse 20q13.2. Rozvoj ochorenia je spojený s nedostatkom proteínu viažuceho guanínový nukleotid (Gs proteín). Súčasne PTH, viažuci sa na receptory cieľových tkanív, nie je schopný aktivovať cyklický adenozínmonofosfát (cAMP) a spôsobiť tkanivovú odpoveď. Pravdepodobne podobný mechanizmus je základom rozvoja necitlivosti tkanív iných orgánov a žliaz s vnútornou sekréciou (hypofunkcia štítnej žľazy, pohlavných žliaz, hypofýzy, diabetes mellitus, ako aj znížená odpoveď pečene na podanie glukagónu), pozorovaná pri pseudohypoparatyreóze typ 1A. Pri tomto type patológie sa normálne zvýšené vylučovanie cAMP do moču v reakcii na exogénne podávanie PTH nepozoruje. Ochorenie je diagnostikované častejšie vo veku 5-10 rokov. Pacienti majú nízky vzrast, krátky krk, okrúhlu tvár, skrátenie záprstných a metatarzálnych kostí (zvyčajne skrátenie štvrtého a menej často druhého prsta) – tzv. brachy-metafalangizmus. Zaznamenáva sa kalcifikácia mäkkých tkanív a podkožné kalcifikácie, ktoré možno zistiť pri narodení; často sa pozoruje súčasné postihnutie ďalších žliaz s vnútornou sekréciou: štítnej žľazy (hypofunkcia), pohlavných žliaz, pankreasu (diabetes mellitus). V dôsledku hypokalcémie často vzniká šedý zákal a defekty zubnej skloviny. Ako diferenciálny diagnostický test na rozlíšenie PHP typu 1A od hypoparatyreózy: nedostatok klinického účinku od parenterálne podanie PTH vo forme zvýšenia hladín vápnika v krvi a zvýšenej renálnej exkrécie fosforu v moči (fosfaturický účinok).
O biochemický výskum Zisťuje sa hypokalciémia, hyperfosfatémia, zvýšená hladina parathormónu v krvi a hypofosfatúria. Hladina Gs proteínu v krvi je znížená. O Röntgenové vyšetrenie Kostrový systém odhaľuje skrátenie záprstných a metatarzálnych kostí, generalizovanú demineralizáciu a zhrubnutie kostí lebečnej klenby.
PSEUDOHYPOPARATROÓZA TYPU 1B
má autozomálne dominantný typ dedičnosti, ale nie je vylúčený dominantný, X-viazaný typ dedičnosti. Je potrebné mať na pamäti niekedy pozorovanú neúplnú penetráciu génu choroby a možnosť latentného nosiča patológie. Preto sa odporúča klinické (zistenie subklinického priebehu ochorenia) a biochemické vyšetrenie (stanovenie hladiny vápnika, fosforu, PTH v krvi) u podozrivých nosičov ochorenia. PHP typu 1B je spôsobené nedostatkom tkanivových receptorov pre parathormón v cieľových orgánoch a obmedzenou rezistenciou na parathormón. Klinický obraz je podobný ako pri type 1A, nedochádza však k poškodeniu iných žliaz s vnútornou sekréciou, menej častá je osteodystrofia.
Pacienti nemajú renálnu odpoveď na exogénne podanie parathormónu vo forme zvýšenej exkrécie cyklického adenozínmonofosfátu v moči, avšak na rozdiel od typu 1A je hladina Gs proteínu v krvi v norme. Ženy sú postihnuté častejšie ako muži, ale závažnosť ochorenia môže byť rovnaká u mužov aj žien.
PSEUDOHYPOPARATHRÓZA TYPU 1C
niektorí autori ho stotožňujú s pseudo-pseudohypoparatyreoidizmom (PPHP), ktorý opísal Albright F. v roku 1952. Vyznačuje sa klinickým obrazom charakteristickým pre PHP, ale hladiny vápnika a fosforu v krvi a moči zostávajú v medziach normy. Hladiny PTH a Gs proteínu v krvi tiež zostávajú na normálnych úrovniach. Niektorí pacienti s PHP typu 1C majú delécie de novo na chromozóme 2. Je možné, že tento variant ochorenia je podtypom PGP typu 1A.
PSEUDOHYPOPARATHRÓZA TYPU 2
klinicky podobné iným typom ochorenia, ale má autozomálne recesívny spôsob dedičnosti. Nemožno vylúčiť existenciu autozomálne dominantných foriem patológie. Vývojová patogenéza je spojená s vnútrobunkovou rezistenciou na cAMP. PTH sa potom viaže na receptory a spôsobuje normálnu bunkovú odpoveď na PTH vo forme zvýšeného vylučovania cAMP. Intracelulárna necitlivosť na cAMP však neumožňuje realizovať plný účinok PTH. Zároveň sa udržiava normálna reakcia obličiek na exogénne podanie parathormónu vo forme zvýšeného vylučovania cyklického adenozínmonofosfátu močom. Bolo navrhnuté, že PHP typu 2 môže súvisieť s nedostatkom vitamínu D.
Identifikované typy PGP sa teda klinicky vyznačujú zníženou citlivosťou cieľových orgánov na PTH, líšia sa však patogenetickými mechanizmami vzniku tkanivovej necitlivosti.
DIAGNOSTIKA
Laboratórnym diferenciálnym diagnostickým testom môže byť vzor renálnej exkrécie cAMP v reakcii na podanie PTH: zvýšené vylučovanie cAMP je pozorované u typu 2 a jeho absencia u typu 1. Diagnóza je potvrdená detekciou zníženej hladiny guanínnukleotidu väzbový proteín (Gs proteín) v krvi (v priemere 1,5–2 krát) v porovnaní s normou. Hypokalciémia sa zvyčajne kombinuje s hyperfosfatémiou a hypofosfatúriou. hladiny PTH sú zvýšené; u typu 1C je hladina PTH normálna, čo vedie k názvu „pseudohypoparatyreóza“. Röntgenovým vyšetrením kostrového systému sa zistí skrátenie záprstných a metatarzálnych kostí, často generalizovaná demineralizácia (osteoporóza), zhrubnutie kostí lebečnej klenby. Dermatoglyfický vzor ukazuje posun axiálneho palmárneho triradia.
Kritériá diagnózy:
- nízky vzrast;
- okrúhla tvár;
- oneskorený neuropsychický vývoj;
- abnormality kostry;
- nízka hladina vápnika v sére;
- vysoká hladina parathormónu v krvi;
- znížené vylučovanie fosfátov a cAMP močom.
LIEČBA A PREVENCIA
Liečba hypokalcémie spočíva v predpisovaní doplnkov vápnika v dávkach dostatočných na udržanie normálnej koncentrácie vápnika v krvi. Veľký význam má terapiu vitamínom D. V súčasnosti sa používajú aktívne metabolity vitamínu D - oxidevit, 1-alfa-D3, kalcitrín atď.v dávke 1–2 mcg/deň s. pozitívny výsledok(zvýšená hladina vápnika v krvi, znížené prejavy konvulzívneho syndrómu). Účinný je aj tachistín (0,5–1,5 mg/deň). Tento liek zvyšuje vstrebávanie vápnika v črevách a tým pomáha zvyšovať hladinu vápnika v krvi. Ako doplnková liečba sa používa antikonvulzívna liečba. Zapnuté intelektuálny rozvoj liečba nemá výrazný účinok, ale spolu s poklesom symptómov konvulzívneho syndrómu sa pozoruje regresia neurologických prejavov (subkortikálne poruchy, choreiformná hyperkinéza, atetóza atď.). Aby sa predišlo predávkovaniu preparátmi vitamínu D, je potrebné sledovať koncentráciu vápnika v krvi každé 3-7 dní počas prvých 2 týždňov liečby a každý mesiac počas nasledujúcich 2-3 mesiacov. Po dosiahnutí stabilnej koncentrácie vápnika v krvi ju stačí kontrolovať raz za 2–3 mesiace. Diéta s obmedzeným obsahom fosforu pomáha normalizovať hladinu fosforu aj vápnika v krvi a zmierňuje príznaky sekundárna hyperparatyreóza. V prípade nedostatočnosti iných endokrinných žliaz, substitučná liečba zodpovedajúce hormóny.
Liečba parathormónom je neúčinná. Na zmiernenie konvulzívnych záchvatov sa intravenózne podáva 10% roztok chloridu vápenatého alebo glukonátu vápenatého; perorálne – 5–10 % roztok chloridu vápenatého, 1 polievková lyžica 3–4 krát denne: glukonát vápenatý, laktát vápenatý – do 10 g denne.
PREDPOVEĎ pre život je určená závažnosťou konvulzívneho syndrómu.
PREVENCIA ochorenie je založené na údajoch z lekárskeho genetického poradenstva.
LEKÁRSKE-GENETICKÉ PORADENSTVO
Pri vykonávaní lekárskeho genetického poradenstva treba vychádzať z autozomálne dominantného typu dedičnosti a vysokého (50%) rizika recidívy ochorenia v rodine s dedičnými formami. Aby bolo možné identifikovať povahu typu dedičnosti, je potrebné vykonať dôkladné vyšetrenie rodičov, pretože syndróm sa môže prejaviť minimálne klinické príznaky. V súčasnosti je vyvinutá a zdokonaľovaná molekulárno-genetická diagnostika ochorenia typizáciou mutácií v géne GNAS1 na chromozóme 20. Vyvíjajú sa metódy prenatálnej diagnostiky ochorenia všeobecne a jeho jednotlivých typov.
KLINICKÉ POZOROVANIE Chlapec G., 14,5 roka (foto 4), bol prijatý do Výskumného klinického ústavu detských chorôb s diagnózou degeneratívne ochorenie. nervový systém? vrodený vonkajší hydrocefalus; symptomatická epilepsia; dedičný syndróm? skladovacia choroba? metabolická encefalopatia; subklinická hypotyreóza; nízkeho vzrastu zmiešaný pôvod; kognitívne poruchy.
Sťažnosti pri príjme k intenzívnym záchvatovitým bolestiam hlavy lokalizovaným vo frontálnej oblasti a sprevádzaným vracaním, ktoré prináša úľavu, zníženú pamäť a výkonnosť v škole, kŕčovité záchvaty, pri ktorých dochádza k zášklbom v pravej ruke.
Fotografia 4.
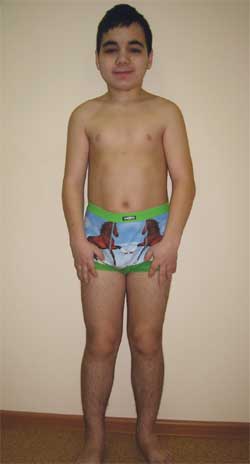
Dieťa G., 14,5 roka, s Albrightovou osteodystrofiou
(fenotypové znaky, nízky vzrast, skrátené končatiny, brachydaktýlia)
Rodinná história: rodičia sú Arméni podľa národnosti, nie sú pokrvne príbuzní a nemajú žiadne profesionálne riziká. V rodokmeni neboli žiadne prípady duševných chorôb, epilepsie alebo oneskorenia vo vývoji. Sibs, sestra, 17-ročná, je vraj zdravá.
História života a choroby: chlapček z 2. tehotenstva, ktoré prebehlo bez zvláštností, druhý pôrod, v termíne, fyziologický, pôrodná hmotnosť - 3100 g, dĺžka - 51 cm, okamžite vykríkol, skóre Apgar - 7/9 bodov. Zhoršenie stavu na 3. deň – novorodenecké kŕče, zastavené v pôrodnici. Skoré postnatálne obdobie je bez rysov. V prvom roku života došlo k miernemu oneskoreniu vo vývoji motoriky, samostatná chôdza od 1 roka 3 mesiacov. V súvislosti s tým bol u neurológa pozorovaný s diagnózou organického poškodenia centrálneho nervového systému; vrodený hydrocefalus; novorodenecké záchvaty; anamnéza febrilných kŕčov.
Dostal som diakarb a finlepsin. Nástup záchvatov po 1 roku 11 mesiacoch. – asymetrické, tonikum vo forme napätia pravá ruka a nohy, s otvorením očí, do 2 minút, bez straty vedomia, časté do 10 epizód denne. Depakine som dostávala nepravidelne. Na pozadí nezávislého stiahnutia - jediný tonický stav. V 2 rokoch bolo urobené CT mozgu v mieste bydliska, kde izolované ohniská demyelinizácia v okcipitálne laloky.
Bol konzultovaný neurochirurg a odporúčaná konzervatívna liečba. Od 3. roku veku dochádza k oneskoreniu psycho-rečového vývinu, odporúča sa pozorovanie u psychiatra.
Od 4. – 5. roku života si rodičia začali všímať deformácie a skracovanie prstov na rukách a nohách, najmä 2. – 4. prstov symetricky na rukách a nohách, a pokles ukazovateľov rastu. Vo veku 8 rokov je záver logopéda o celkovej poruche reči 2. – 3. stupňa, odporúča sa vzdelávanie v špecializovanej škole. V rovnakom veku vyšetrenie genetikom v mieste bydliska, záver: dedičné ochorenie výmena? Odporúčala sa štúdia aminokyselín v krvi, nezistili sa žiadne zmeny; konečný záver: nezistil sa žiadny dôkaz dedičného metabolického ochorenia; hypochondroplázia; Odporúča sa liečba neurológom a endokrinológom.
Tabuľka.
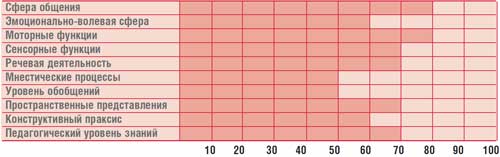
Profil duševného vývoja dieťaťa G., 14,5 ročného (IQ = 68)
Vo veku 8 rokov bol konzultovaný endokrinológom ohľadom oneskoreného rastu a vývoja. Röntgenové vyšetrenie rúk odhalilo tieto znaky: stredné, hlavné falangy a záprstné kosti skrátený, zahustený; Diagnóza rádiológa znela achondroplázia.
Opakovane vyšetrený v mieste bydliska v neurologickej nemocnici. Vo veku 12 rokov sa objavili kŕčové záchvaty bez straty vedomia so zášklbmi pravej ruky, ktoré boli sériového charakteru, bola predpísaná antikonvulzívna liečba (Depakine), frekvencia záchvatov sa výrazne znížila. V 13 rokoch bola vykonaná MRI mozgu s kontrastom - symetrické zmeny v báze temporálnych lalokov na jadrovej úrovni v podobe zvýšenia signálu MR, ktorý je typický pre toxické (mangánové) alebo metabolické (meď, železo) encefalopatie.
Opakované vyšetrenie vo veku 13 rokov 3 mesiace. endokrinológ, štúdia profilu štítnej žľazy odhalila zvýšenie hormón stimulujúci štítnu žľazu(TSH), diagnostikovaná subklinická hypotyreóza, predpísaný L-tyroxín.
Pri analýze ambulantnej karty a dokumentácie dieťaťa v mieste bydliska bola raz vykonaná štúdia vápnika a fosforu, vo veku 1,5 roka bola zaznamenaná hypokalcémia, ale pri tejto príležitosti sa nevykonalo žiadne ďalšie vyšetrenie. Vzhľadom na neistotu diagnózy v mieste bydliska poslal genetik dieťa do Moskvy, do Vedecko-výskumného klinického ústavu pediatrie, aby diagnózu objasnil.
Údaje z objektívneho výskumu:
Výška – 143 cm, hmotnosť – 43 kg.
Fyzický vývoj veľmi nízka, harmonická, disproporčná stavba tela v dôsledku skrátených končatín. Rast Sds zodpovedá –2,8 odchýlkam od normy (norma –2+2).
Fenotypové znaky: okrúhla tvár, krátky krk, antimongoloidné palpebrálne štrbiny, široký nosový mostík, vysoké čelo, brachydaktýlia, skrátenie IV a V záprstných a metatarzálnych kostí (foto 5). Vnútorné orgány – bez akýchkoľvek zvláštností. Sexuálny vývoj – III-IV štádium tannera (čo zodpovedá veku).
Laboratórne a funkčné výskumné údaje:
Klinická analýza krv a moč sú normálne.
Biochemický krvný test: celkový vápnik – 1,39 (normálne 2,02–2,6 mmol/l), ionizovaný vápnik – 0,61 (normálny 1,13–1,32 mmol/l), anorganický fosfor – 3,66 (normálne 0,86–1,56 mmol/l), ostatné ukazovatele sú v rámci normálne limity.
Biochemický rozbor moču: vylučovanie fosfátov obličkami je znížené – 11,5 mmol/l (normálne 19–32 mmol/l).
Profil štítnej žľazy: TSH – 11,75 (normálny rozsah 0,4–4,0 µIU/ml), voľný T4 – 0,49 (normálny rozsah 1,0–1,8 ng/dl).
Parathormón – 499 (normálne 12–65 pg/ml), STH – 7 ng/ml (normálne 7–10 ng/ml), somatomedín-C – 250 ng/ml (normálne 88–360 ng/ml).
Ultrazvuk vnútorných orgánov - bez akýchkoľvek funkcií.
EKG – migrácia supraventrikulárneho kardiostimulátora na pozadí pravidelnej srdcovej frekvencie 71–80 úderov/min. Neúplná blokáda pravá noha Jeho zväzok. Porušenie procesu repolarizácie v myokarde zadnej steny ľavej komory (pokles ZT III, aVF).
R-grafia chrbtice – pravostranná skolióza hrudný chrbtice 1. stupňa, ťažká osteoporóza.
R-grafia rúk so zachytením predlaktí - skrátenie a rozšírenie koncových a stredných falangov. Kostný vek - 13,5-14 rokov.
Neboli zaznamenané žiadne EEG vzory epileptickej aktivity.
MRI mozgu - MR obraz mnohopočetných subkortikálnych ložísk zvýšeného MR signálu v čelné laloky, vonkajší kompenzovaný hydrocefalus s atrofiou mozgovej substancie.
MSCT mozgu ukazuje symetrické oblasti kalcifikácie lentiformných jadier. Difúzne hyperdenzné oblasti v talami, kaudátové jadrá s oblasťou kalcifikácie vpravo. Viacnásobné bodové kalcifikácie vnútorných mäkkých tkanív lebky.
Audiogram – bez patológie.
Prebieha diagnostika DNA v géne GNAS1.
Odborné konzultácie:
Endokrinológ – Albrightova dedičná osteodystrofia typu 1A (pseudohypoparatyreóza). Primárna hypotyreóza, neúplná kompenzácia liekov.
Oftalmológ – úplná sekundárna katarakta. Odporúčané chirurgická liečba.
Psychológ – kognitívna porucha (psychologický profil dieťaťa je uvedený v tabuľke).
Berúc do úvahy fenotyp dieťaťa, anamnézu, výsledky dodatočných štúdií (hypokalcémia, hyperfosfatémia, hypofosfatúria, zvýšená hladina parathormónu v krvi), kalcifikácie v mozgu, prítomnosť katarakty, hypotyreóza) bola stanovená diagnóza: Albrightova dedičná osteodystrofia typu 1A (pseudohypoparatyreóza). Odporúča sa vykonať DNA diagnostiku – hľadanie mutácií v géne GNAS1.
Liečba: Dieťaťu sa odporúča užívať eutirox v dávke 100 mcg/deň; aktívny metabolit vitamínu D - alfa-D3 (Teva) v dávke 2 mcg/deň; vápnik (Sandoz) 2000 mg/deň; neustále používanie antikonvulzívnej terapie - finlepsin 800 mg / deň pod dohľadom neurológa-epiptológa; triedy s rečovým patológom a psychológom; energeticko-tropná terapia (Elkar a koenzým Q10 v dávkach súvisiacich s vekom). Sledovanie ukazovateľov metabolizmu fosforu a vápnika, hladiny parathormónu.
teda Prezentované klinické pozorovanie demonštruje náročnosť diferenciálnej diagnostiky, dôležitosť včasného štúdia jednoduchých biochemických parametrov (v prípade epilepsie je povinný opakovaný skríning ukazovateľov metabolizmu fosforu a vápnika), výsledky neskorej diagnostiky geneticky podmieneného ochorenia. , potreba integrovať jednotlivé znaky do celkového fenotypu konkrétneho ochorenia patologický stav na cielenú a včasnú diagnostiku samostatné formuláre dedičné choroby. Včasná diagnostika a objasnenie genézy každého syndrómu sú obzvlášť dôležité, pretože nám umožňujú nájsť optimálny prístup k liečbe týchto stavov a prevencii možné komplikácie(až do zdravotného postihnutia dieťaťa); prevencia recidívy dedičných chorôb v postihnutých rodinách (lekárske a genetické poradenstvo). To diktuje potrebu lekárov rôznych špecializácií jasne navigovať tok dedične podmienenej patológie. Zoznam referencií je v redakcii.

