Arnold-Chiari е вродена или аномалия, която възниква по време на формирането на дете в утробата. Аномалията възниква поради компресия на мозъка, което причинява деформация на черепните области. Последствията са: малкия мозък и мозъчен стволсилно се разместват и падат в тилна част, производителността е нарушена.
Всички процедури трябва да се извършват стриктно според препоръките на лекаря. Истински специалист няма да сложи точна диагнозабез извършване на подходящ преглед.
Ефективно лечение на синдрома
В момента се използват два вида лечение: хирургично, когато става дума, и консервативно.

Консервативното лечение се използва, когато заболяването не причинява силен дискомфорт на пациента и не засяга неговото развитие. Лекарят препоръчва по-често да правите физически упражнения и упражнения за мускулна координация. Предписват се и някои лекарства: болкоуспокояващи, мускулни релаксанти, противовъзпалителни средства. Освен това се предписва комплекс, особено група В, тъй като те са отговорни за биохимичните процеси в организма и нормализират функционирането на централната нервна система.
Разбира се, такива рецепти няма да помогнат напълно да се отървете от болестта, но ще ви позволят да правите без операция възможно най-дълго.
Ако малформацията на заболяването прогресира, тогава ще е необходимо спешно лечение. хирургична интервенция. Извършва се операция или байпас. Операцията решава две основни причини:
- Коригирайте дефекти, които допринасят за компресия на черепа и мозъка.
- Възстановява нормалното движение на цереброспиналната течност.
Тази операция е доста често срещана и продължава не повече от два часа. Пациентът се възстановява напълно в рамките на няколко седмици. Благодарение на операцията вътречерепното налягане се нормализира, а пространството в гръбначния и главния мозък се увеличава, болестта отстъпва.
Предпазни мерки
Винаги трябва да се грижите за здравето си, а ако това е период, когато жената е бременна под сърцето си, тогава отговорността се удвоява. Има някои превантивни мерки за предотвратяване на заболяването:
- включете повече плодове и зеленчуци в диетата си
- пийте пресни сокове, яжте млечни продукти и меса, богати на протеини
- вземете пренатални витамини
- предавам се лоши навици, ако има такива
- приемайте само онези лекарства, които са допустими по време на бременност и само според предписанието на лекар
- извършват всички необходими изследвания
Ако следите диетата си и се храните добре, здрав образживот, вземете тестове навреме и слушайте лекаря, тогава вероятността от раждане на здраво дете се увеличава многократно.
Така че синдромът на Арнолд-Киари в плода възниква поради различни причини, както вродени, така и придобити. Болестите от тип 1 и 2 са напълно лечими, ако се извърши необходимата операция. За да се предотврати появата на патология, на бъдещата майканеобходимо е да се грижите максимално за детето си, като по този начин това ще има положителен ефект върху развитието на мозъка на нейния плод.
25 февруари 2017 г Виолета доктор
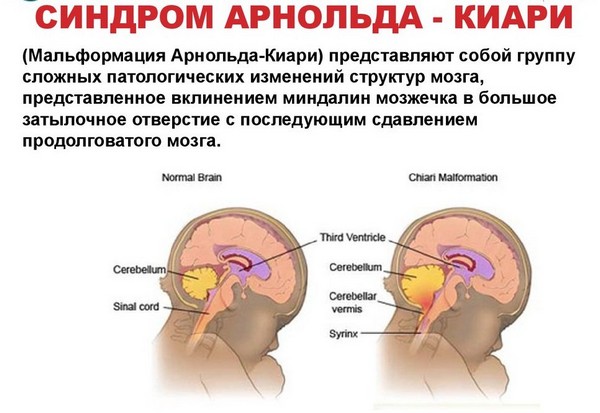
Малформацията на Арнолд-Киари е малформация на цервико-медуларната връзка, характеризираща се с изместване на церебеларните тонзили и в някои случаи на ствола и IV вентрикула под нивото на foramen magnum.
J. Cleland е първият, който описва малформации през 1883 г. При аутопсията той разкрива удължаване на багажника и спускане на церебеларните тонзили в гръбначния канал при 9 мъртви бебета. Работата на J. Cleland обаче не беше забелязана. Тогава през 1891 г. Hans von Chiari описва вродена аномалия, състояща се от хернизирана изпъкналост на церебеларните тонзили под нивото на foramen magnum.
Видове малформации
Киари малформация тип I- изместване на маломозъчните тонзили надолу през foramen magnum към горните части гръбначен мозък. Този тип малформация е придружена от хидромиелия и обикновено се появява в юношеска или зряла възраст. При подрастващите основните симптоми са нарушена флексия и намалена сила в ръцете, загуба на болкова и температурна чувствителност в горната половина на тялото и ръцете. Възрастните обикновено се оплакват от болка в шийката на матката тилната област, засилваща се при кашлица, както и болки в ръцете.
Киари малформация тип IIхарактеризиращ се с изместване на вермиса на малкия мозък, сливиците, четвъртата камера и продълговатия мозък(части от мозъчния ствол) във foramen magnum. Този тип, наричан още малформация на Arnold-Chiari, е много по-често придружен от хидромиелия, отколкото тип I и почти винаги е свързан с миеломенингоцеле. Миеломенингоцеле е вродено нарушение на затварянето на гръбначния мозък и гръбначния стълб по време на формирането на плода. Симптомите на тази малформация са очевидни и обикновено се появяват веднага след раждането заедно с кратки епизоди на спиране на дишането, намален фарингеален рефлекс, неволеви и бързи движения очни ябълкинадолу, намалявайки силата в ръцете.
Киари малформация тип IIIсе състои от изместване на малкия мозък и част от мозъчния ствол с менингите в менингоцеле, разположено в цервико-тилната област.
Преди това беше идентифицирана малформация на Chiari тип IV, която е придружена от недоразвитие на малкия мозък. Понастоящем обаче повечето автори предпочитат да класифицират този тип дефект в развитието като аномалия на Денди-Уокър.
Малформациите на Арнолд-Киари от тип II и III могат да бъдат придружени от признаци на дисплазия на нервната система: полимикрогирия, хетеротопия на кората, хипоплазия на субкортикалните ганглии, дисгенезия на corpus callosum, патология на септум пелуцидум, удебеляване на интерталамуса кръстовище, клюновиден тектум (coracoid tectum), често се отбелязва наличието на инфлексия на Силвиевия акведукт (55%), кисти на отвора на Magendie, хипоплазия на фалкс и тенториум на малкия мозък, полупрешлени, ниско местоположение на каудалния част от гръбначния мозък на нивото на LIV–V прешлени и по-долу.
Етиология на малформацията на Киари
Етиологията на заболяването към момента е неясна. Има доказателства за роля генетичен факторв етиологията на този синдром. Ектопични церебеларни тонзили във foramen magnum бяха открити при три монозиготни близнаци. След първото описание на малформацията на Cleland през 1883 г. се появиха няколко теории. Теорията, подкрепена от проучванията на Misao Nishikawa et al., е, че поради параксиална дисплазия на мезодермалния слой или първично увреждане на структурите на съответния сомит се образува необичайно малка задна черепна ямка, структурите на задния мозък, изпълващи обем на задната черепна ямка и продължавайки да расте, се спускат в тилния канал. Комбинацията от малформация на Киари тип II с менингомиелоцеле се дължи на факта, че степента на параксиална дисплазия на мезодермалния слой при тип AK - II е по-изразена, отколкото при тип AK - I и се отбелязва не само на нивото на тилната кост, формация, но и по оста на тялото на ниво редообразуващи прешлени, което се проявява в спина бифида, както и в аномалии на редица други костни структуриИ скелетна системав общи линии.
Клинична картина на малформация на Киари
Клинични проявления АК – тип Iсе появяват най-често в юношеска или зряла възраст. Тези прояви се вписват в такива неврологични синдроми като церебелобулбарен, ликворохипертензивен, сирингомиеличен, синдром на нараняване черепномозъчни нерви. CSF хипертензивен синдром се проявява с главоболие, обикновено субокципитално, и болка във врата, влошена от кашлица, кихане и напрежение, застояли дисковезрителни нерви. Стволовите нарушения и нарушенията на функциите на черепните нерви се проявяват под формата на нестабилна осцилопсия, тригеминална дизестезия, загуба на слуха, шум в ушите, замаяност, дисфагия, спиране на дишането по време на сън, периодично припадане (често свързано с кашлица), нарушен контрол върху сърдечната честота кръвно налягане при преминаване от хоризонтално във вертикално положение, може да се наблюдава атрофия на половината език и парализа гласни струни, стридор, спастична или комбинирана (повече в горните крайници) тетрапареза. Церебеларни нарушения- нистагъм, дизартрия, атаксия. Симптомите, свързани със сирингомиеличните кисти, са изтръпване, нарушение на чувствителността, обикновено от дисоцииран тип, както и невроартропатия, дисфункция на тазовите органи, липса на коремни рефлекси, мускулна загуба. В същото време редица автори отбелязват несъответствие между локализацията, степента на кистата, кистозния индекс (съотношението на предно-задния размер на кистата към диаметъра на гръбначния мозък на нивото на кистата), от една страна, и зоната на хипестезия, разпространението на сегментарни нарушения на повърхностната чувствителност, тежестта на мускулната загуба и степента на пареза - от друга.
АК тип IIсе проявява при новородени и рано детствосимптоми като апнея, стридор, двустранна пареза на гласните струни, неврогенна дисфагия с назална регургитация, цианоза по време на хранене, нистагъм, хипотония, слабост, спастичност в горните крайници, която може да прогресира до тетраплегия.
Киари III малформациятип е рядък, клиничните му прояви са същите като при АК II.
Диагностика на малформация на Киари
Стандартен рентгеново изследванеможе да разкрие само индиректни признаци на AC малформация, компютърна томографиясъщо не осигурява ясна визуализация на мекотъканните структури. Широкото въвеждане на ЯМР в клиничната практика реши повечето от проблемите, свързани с диагностицирането на малформацията на Chiari. Това беше улеснено от добрата визуализация на структурите на задната черепна ямка, краниовертебралната връзка, гръбначния мозък и липсата на артефакти от костните структури.
Лечение на малформация на Chiari
Възможно е само лечение на малформация на Chiari и съпътстваща сирингомиелия хирургично. Операцията се състои в декомпресия на задната черепна ямка или инсталиране на шънт за цереброспинална течност за съпътстваща хидроцефалия.
Извършва се локална декомпресия под обща анестезияи се състои в отстраняване на част от тилната кост, както и на задните половини на I и/или II шиен прешлен до мястото, където се спускат малкомозъчните тонзили. Спуснатите церебеларни тонзили също се резецират, като по този начин се елиминира компресията на мозъчния ствол. Това ефективна работаразширява foramen magnum и облекчава компресията на мозъчния ствол, гръбначния мозък и сливиците на малкия мозък. Операцията също така отваря твърдата мозъчна обвивка, дебелата мембрана, която обгражда мозъка и гръбначния мозък. В отворената твърда мозъчна обвивка се зашива пластир от друга тъкан (изкуствена или взета от пациента) за по-свободно преминаване на цереброспиналната течност.
По-рядко се извършват дренажни операции гръбначно-мозъчна течностот разширения гръбначен мозък до гръдния или коремна кухинас помощта на специален кух шунт с клапа или в интратекалното пространство. Понякога тези операции се извършват на етапи.
Ефективността на хирургичната декомпресия варира от 50 до 85% според различни автори. Трябва да се помни, че хирургичното лечение се извършва най-добре преди развитието на тежки неврологични разстройства, тъй като след хирургично лечениефункционалното възстановяване не настъпва напълно или изобщо не настъпва и основната цел на операцията е да стабилизира неврологичния статус на пациента и да предотврати по-нататъшното прогресиране на заболяването.
Израелският център по неврохирургия и неврология Neuromed е специализиран в неврохирургични операции за малформация на Arnold-Chiari, използвайки иновативни техники, което позволява да се повиши ефективността и безопасността на лечението.
Характеризира се с несъответствие между размерите на задната черепна ямка и разположените в тази област мозъчни структури, в резултат на което малкомозъчните тонзили и мозъчният ствол се спускат във foramen magnum. Това води до тяхното нарушение в тази област. Често тази патологиясъчетано с нарушения в развитието на гръбначния мозък.
Повечето пациенти със синдром на Арнолд-Киари не изпитват никакви симптоми и заболяването най-често се открива случайно, по време на диагностиката на други заболявания. Малформацията на Chiari е рядка: засяга приблизително 3-8 души на сто хиляди случая.
Какви са причините за синдрома на Arnold-Chiari?
Към днешна дата етиологията на това заболяване не е напълно проучена и експертите нямат консенсус относно причините за заболяването. Има предположение, че малформацията на Arnold-Chiari е причинена от структурни аномалии в мозъка и гръбначния мозък. Те се развиват в периода на вътрематочно развитие и са свързани с генетични мутацииили небалансирана диета на жена по време на бременност, когато в диетата липсват хранителни вещества.
Според друга версия, малформацията на Chiari възниква поради увеличения размер на мозъка и изглежда измества съдържанието на задната черепна ямка през foramen magnum.
Преход на незначителен дефект в изразен клинична формаможе да причини хидроцефалия. Причинява се от увеличаване на обема на мозъка поради разширяване на вентрикулите. Поради факта, че със синдрома на Chiari, в допълнение към дисплазията костна тъканкраниовертебрална връзка, има недоразвитие лигаментен апаратв тази област дори най-леката травматична мозъчна травма може да влоши проникването на малкия мозък на тонзилите във foramen magnum.
Класификация на малформацията на Arnold-Chiari
Тази патология е разделена на четири вида:
- Малформация на Арнолд-Киари I степен- проявява се със слизане на малкомозъчните тонзили под нивото на foramen magnum. По правило се проявява в юношеството и по-късните години. Често придружен от хидромиелия, при която цереброспиналната течност се натрупва в гръбначния мозък, в централния му канал;
- Малформация на Arnold-Chiari II степен- се проявява почти веднага след раждането на бебето, в първите дни от живота му. При тази форма на заболяването през foramen magnum, освен сливиците, излизат и продълговатият мозък, вермисът на малкия мозък и четвъртата камера. Този тип заболяване често се комбинира с хидромиелия и почти винаги се свързва с наличието на вродена спинална херния;
- Малформация на Arnold-Chiari III степен- проявява се от факта, че малкият мозък и продълговатият мозък, спускащи се под foramen magnum, се намират в менингоцеле на цервико-тилната област;
- Малформация на Arnold-Chiari IV степен- проявява се с недоразвитие (хипоплазия) на малкия мозък, при което няма изместване в каудална посока. Някои експерти приписват този дефект на синдрома на Dandy-Walker, когато в допълнение към хипоплазията на малкия мозък има вродени кисти на задната черепна ямка и хиперцефалия.
Киари малформации от степен I и II често се комбинират с различни патологиинервна система, като например:
- полимикрогирия;
- хетеротопия на мозъчната кора;
- дефекти на corpus callosum;
- кисти на отвора на Maugendie;
- хипоплазия на субкортикалните структури;
- извивка на Силвиев акведукт и др.
Малформация на Арнолд-Киари: симптоми
Симптомите на малформация на Arnold-Chiari варират в зависимост от вида на заболяването. IN клинична практикаНай-честата е малформация на Chiari тип I, при която се засягат черепномозъчните нерви, наблюдават се и синдроми на сирингомиелична, церебелобулбарна и ликворна хипертония. Този тип заболяване обикновено се открива в зряла възраст или по време на пубертета.
Ликворно-хипертензивният синдром, който придружава малформацията на Chiari I, се характеризира с главоболие в тилната и цервикалната област. Болезнени усещаниявлошават се, когато мускулите на врата са напрегнати, например при кашляне или кихане. Често се появява повръщане, което няма нищо общо с приема на храна и нейния характер. При преглед се установява повишен тонус на мускулите на врата. Церебеларни нарушенияпроявява се с дизартрия (неясен говор), неволеви движенияи двигателно разстройство.
Увреждането на мозъчния ствол и разположените в него коренчета и ядра на черепните нерви причинява замъглено виждане, несвързан говор, нарушение на гълтането, загуба на слуха, системно замаяност с усещане за въртящи се предмети, шум в ушите, синдром на сънна апнея, повтарящи се краткосрочни загубисъзнание, ортостатичен колапс. Пациентите отбелязват повишен шум в ушите и замайване при завъртане на главата, което може дори да причини припадък. Възможна е пареза на ларинкса и затруднено дишане.
Малформациите на Arnold-Chiari от степен II и III са сходни по отношение на техните клинични проявления. Те стават забележими още в първите дни от живота на бебето. Малформацията на Chiari II се характеризира с шумно дишане, периоди на кратко спиране на дишането, невропатична ларингеална пареза и проблеми с преглъщането, при които течната храна се връща обратно в носа. При новородени се наблюдава нистагъм, повишен мускулен тонус, цианоза на кожата по време на хранене. Може също да присъства двигателни нарушенияв различна степен.
Малформацията на Chiari III е по-тежка форма на заболяването и често развитието на плода е несъвместимо с живота.
Как се диагностицира синдромът на Arnold-Chiari?
При диагностицирането на тази патология нито едното, нито другото медицински преглед, нито наборът (РЕГ, ЕЕГ, Ехо-ЕГ) не дават данни, с които да се постави точна диагноза. Те разкриват само признаци на хидроцефалия.
Рентгеновото изследване открива само костните дефекти, придружаващи малформацията на Chiari. Преди въвеждането на томография в неврологична практикадиагностика на това заболяванебеше трудно. Сега може да се постави точна диагноза.
Синдром на Арнолд-Киари: лечение
Както беше отбелязано по-горе, заболяването в повечето случаи протича без никакви симптоми, така че много често синдромът на Арнолд-Киари не изисква лечение. Ако има болка в цервикалната и тилната област, се провежда консервативна терапия. Той включва аналгетици, мускулни релаксанти и противовъзпалителни лекарства.
Неврологични разстройства (нарушения на мускулния тонус и чувствителност, парези и др.), Както и в случай на неефективно лечение на болкови синдроми консервативен методизползва се хирургична интервенция, чиято цел е да се спре процесът структурни променив гръбначния стълб и мозъка и стабилизира симптомите. Ако операцията е успешна, налягането върху малкия мозък се намалява и изтичането на цереброспиналната течност се възстановява.
Има заболявания, свързани с ненормално разположение на органите, което причинява дисфункция в тези области. Едно от тези заболявания е синдромът на Арнолд Киари (малформация на Киари). Характеризира се с анормално разположение на малкия мозък и мозъчния ствол в черепа, поради което те леко изпъкват във foramen magnum. Това явление е придружено от болезнени атаки в задната част на главата, несвързана реч, липса на координация, отслабване на мускулите на ларинкса и други симптоми. Заболяването е разделено на 4 вида, всеки от които има свои собствени симптоми и лечение.
Малформацията на Chiari се диагностицира доста просто и всичко, което е необходимо, е MRI. Патологията може да бъде излекувана само чрез операция.
Има определено разделение между гръбначния мозък и главния мозък, което е foramen magnum. В това място те се свързват и над него се намира задната черепна ямка, съдържаща малкия мозък, моста и продълговатия мозък. При болестта на Chiari мозъчната тъкан попада във foramen magnum, който компресира областите на продълговатия мозък и гръбначния мозък, локализирани в тази област. Често поради този процес изтичането на цереброспиналната течност от главата (гръбначно-мозъчната течност) се влошава, което води до развитие на хидроцефалия (хидроцефалия).
Синдромът на Киари се отнася до вродени патологии на ставата на черепа и горна частгръбначен стълб.
Тази диагноза се поставя доста рядко и според статистиката се наблюдава при 10 новородени на 120 хиляди души.
Наличието на заболяването може да се установи в рамките на един или два дни от момента на раждането, но някои видове заболяване се откриват едва в зряла възраст. Най-често болестта на Арнолд Киари се развива заедно със сирингомиелия, която се характеризира с празни участъци в субстанцията на гръбначния мозък и продълговатия мозък.
Причини за развитие
Малформацията на Арнолд Киари все още не е особено проучена и никой не може да назове точните фактори, които влияят върху развитието на болестта. Има много предположения, например някои експерти смятат, че патологията е причинена от твърде малкия размер на задната черепна ямка. В резултат на това тъканите нямат достатъчно място и те се спускат в тилния отвор. Алтернативна теория е, че мозъкът е по-голям от нормалното при раждането. Ето защо тя избутва част от тъканите си в тилния отвор.
Поради стагнацията на цереброспиналната течност и последващото образуване на хидроцефалия, патологията има по-изразени симптоми. В крайна сметка, поради това заболяване, вентрикулите се увеличават и в резултат на това размерът на мозъка. Този процес засилва екструзията на церебеларните тонзили във foramen magnum. Нараняванията на главата също могат да влошат заболяването. Болестта на Киари се характеризира не само с анормално развитие на преходните тъкани между гръбначния и главния мозък, но и с аномалии на лигаментния апарат. Всяко увреждане на областта на главата притиска малкия мозък още повече в тилната област, което води до по-изразено заболяване.
Видове патологичен процес
Малформацията на Chiari има следните видове развитие:
- Първи изглед. Малформация на Арнолд Киари тип 1 се характеризира с навлизане на малкия мозък на тонзилите във форамен магнум. Заболяването се проявява главно в юношеска възраст, а понякога и в зряла възраст. Хидромиелия се среща много често при тип 1 малформация на Arnold Chiari;
- Втори изглед. Тази диагноза се поставя още в първите дни след раждането на детето. Тип 2 малформация на Arnold Chiari е много по-тежка от тип 1. В неговите случаи част от малкия мозък (в допълнение към амигдалата), както и четвъртата камера и продълговатия мозък също влизат в тилната кухина. Хидроцефалия с този вид заболяване се среща много по-често. Причината за това явление в повечето случаи се крие във вродена;
- Трети тип. Разликите му от втория тип са, че тъканите, които се спускат във foramen magnum, завършват в менингоцеле (херния), което се намира в цервико-тилната част;
- Четвърти тип. Същността му се крие в недоразвития малък мозък, който, за разлика от други видове заболяване, не се измества към foramen magnum. Някои експерти смятат, че болестта Киари тип 4 е част от синдрома на Денди-Уокър. Характеризира се с развитието на кисти, локализирани в задната черепна ямка и хидроцеле на мозъка.
Вторият и третият тип аномалия често се срещат в комбинация с неправилно развитиедруги тъкани на нервната система, а именно:
- Атипично разположение на тъканите на кората на главния мозък;
- Патологии на corpus callosum;
- Полимикрогирия (много малки извивки);
- Недоразвитие на тъканите на подкоровите структури, както и на церебеларния фалкс.
Симптоми
Най-често се диагностицира малформация на Арнолд Киари 1 степен. Той съчетава следните синдроми:
- сирингомиелична;
- Ликворна хипертония;
- Церебелобулбарен.
На този фон черепно-мозъчният нервни влакна, и живеят без симптоми с тази патология до юношеството. При някои хора първите признаци стават видими след 20-годишна възраст.
Ликворно-хипертензивен синдром, който е характерен за аномалията на Арнолд Киари, се проявява със следните симптоми:
- Болка в цервикалната и тилната област, която се проявява по време на кашляне и кихане, както и поради мускулно напрежение на това място;
- Безпричинно повръщане;
- Напрежение на мускулите на врата;
- несвързана реч;
- Нарушена координация на движенията;
- Неконтролируемо трептене на очите (нистагъм).
С течение на времето, поради увреждане на мозъчния ствол и околните нерви, човек изпитва следните симптоми:
- Влошаване на зрението;
- Двойно изображение пред очите (диплопия);
- Проблеми с преглъщането;
- Увреждане на слуха;
- Чести световъртежи;
- Шум в ушите;
- Нарушение на съня, придружено от спиране на дишането през носа и устата за 10 секунди или повече;
- Загуба на съзнание;
- Недостатъчно кръвоснабдяване на мозъка, проявяващо се поради ниско кръвно наляганепри промяна на позицията на тялото.
При малформация на Арнолд Киари симптомите се засилват поради внезапни завои на главата и заболяването се проявява, както следва:
- Повишено замаяност;
- Шумът в ушите става по-силен;
- Загуба на съзнание по-често;
- Половината от езика претърпява атрофична промяна (намаляване на размера);
- Мускулите на ларинкса отслабват, което затруднява дишането и гласът става дрезгав;
- Отслабване на мускулите на крайниците, главно на горните.
Често патологията се характеризира със сирингомиеличен синдром и в този случай се проявява, както следва:
- Влошаване на чувствителността;
- Белтъчно-енергиен дефицит (хипотрофия) на мускулната тъкан;
- изтръпване;
- Отслабване или пълна липса на коремни рефлекси;
- тазови нарушения;
- Неврогенна артропатия (деформация на ставите).
Церебелобулбарният синдром, който се проявява при болестта на Киари, се проявява със следните симптоми:
- Дисфункция на тригеминалния нерв;
- Вестибулокохлеарна нервоза;
- Проблеми с координацията;
- нистагъм;
- замаяност
Втората и третата степен на заболяването се проявяват по подобен начин, но първите симптоми са видими в рамките на 2-3 дни от раждането. Тип 2 малформация на Арнолд Киари има свои собствени отличителни черти:
- Силно дишане, което понякога спира за 10-15 секунди;
- Двустранно отслабване на мускулите на ларинкса, което води до проблеми с преглъщането и течната храна често се хвърля в носа;
- нистагъм;
- Втвърдяване на мускулите Горни крайниципоради повишен тон;
- Син в лицето кожата(цианоза);
- Затруднено движение до парализа на по-голямата част от тялото (тетраплегия).
Третият вид аномалия е много по-тежка и рядко е съвместима с живота поради сериозни нарушения.
Диагностика
В старите времена беше изключително трудно да се диагностицира патологията, тъй като разпитът, прегледът и стандартните методи на изследване не дадоха много резултати. Те показаха наличност високо кръвно наляганеи воднянка на мозъка. Рентгеновата снимка малко опрости задачата, тъй като показа костни деформации в черепа, но това не ни позволи да бъдем напълно сигурни в диагнозата. Ситуацията се промени след появата на топографските изследвания. В края на краищата, този диагностичен метод направи възможно пълното изследване на образуванията на задната черепна ямка. Ето защо MRI (магнитен резонанс) се счита за незаменим метод за изследване за диференциране на синдрома на Киари от други патологични процеси.
С помощта на ЯМР ще трябва да изследвате областта на шията и гърдите. Всъщност в тези места на гръбначния стълб често има менингоцеле, както и сирингомиелични кисти. По време на прегледа лекарят трябва не само да се увери, че синдромът на Киари е налице, но и да изключи други патологични процеси, които често се комбинират с него.
Курс на терапия
При малформация на Arnold Chiari лечението не се изисква само ако патологията е асимптоматична. В такава ситуация трябва да се избягват удари и наранявания на главата и шията, за да не се влоши хода на заболяването.
Ако заболяването протича с минимални симптоми, а именно лека болка, тогава е необходим курс от лекарства с аналгетичен и противовъзпалителен ефект. Предписването на мускулни релаксанти за отпускане на мускулите няма да навреди.
IN тежки случаиКогато заболяването протича с изразени симптоми (отслабване на мускулите, дисфункция на черепните нерви и др.), Ще се наложи хирургична интервенция. По време на операцията лекарят ще разшири foramen magnum, като отстрани част от тилната кост. Ако трябва да премахнете натиска от тялото и гръбначния мозък, ще трябва да премахнете част от сливиците на малкия мозък и предната половина на 2-та горни прешлена. За да нормализира циркулацията на цереброспиналната течност, хирургът ще трябва да направи пластир в твърдата мозъчна обвивка.
В някои случаи хирургичното лечение се извършва с помощта на байпас. Тази операция е клон на цереброспиналната течност с помощта на дренаж, в резултат на което тя престава да стагнира.
Прогноза

Много хора, страдащи от синдрома на Арнолд Киари, се чудят колко дълго трябва да живеят. На този въпрос може да се отговори въз основа на вида на патологията и тежестта на курса. Важен факторВажно е и навременността на хирургическата интервенция.
Хората, страдащи от болест на Chiari тип 1, често имат средна продължителност на живота, тъй като патологията може да бъде асимптоматична. При наличие на 1 или 2 вида заболяване неврологични признаци, тогава е важно операцията да се извърши възможно най-скоро. В края на краищата, усложненията, свързани с мозъка и гръбначния мозък, са практически нелечими. Третият вид заболяване най-често завършва със смъртта на пациента при раждането.
Болестта на Арнолд Киари е вродена аномалия, която трябва да се лекува при първите симптоми. В противен случай може да останете трайно инвалид или да загубите живота си.
Малформация на Арнолд-Киарие вродена патология на развитието ромбенцефалон, проявяващо се в несъответствие между размерите на задната черепна ямка и мозъчните структури, разположени в тази област, което води до спускане на мозъчния ствол и церебеларните тонзили във foramen magnum и тяхното нарушаване на това ниво.
В повечето случаи дефектът се комбинира с хидроцефалия и аномалии на гръбначния мозък. Причините могат да бъдат вродена дисплазия (увреждане) на широкия тилен отвор, чиито размери стават значително по-големи от нормалните.
За първи път е описано от N. Chiari през 1896 г. Това състояние се характеризира с каудално изместване на продълговатия мозък, моста и червея на малкия мозък, когато всички тези структури завършват в шийната част на гръбначния стълб.
Честотата на това заболяване варира от 3,3 до 8,2 наблюдения на 100 000 души от населението.
Истинската честота на различни видове синдром на Арнолд-Киари и честотата на този дефект като цяло не е установена. Една от причините за липсата на такива данни са различните подходи към класификацията на този дефект. Според Международна класификациязаболявания, синдромът на Арнолд-Киари има отделен код (Q07.0), но се дефинира в него като „... патологично състояние, при което има увеличение вътречерепно наляганев резултат на интракраниален тумор, оклузивни форми на хидроцефалия, възпалителен процес, който в някои случаи води до херния на малкия мозък и продълговатия мозък във форамен магнум.” В ултразвуковата пренатална литература все още не е възможно да се намерят описания на случаи на пренатална диагностика на синдрома на Arnold-Chiari, които напълно отговарят на тези характеристики.
Морфологичните особености на различни видове дефект на Harold-Chiari определят възможностите за пренатално откриване и прогноза за живота.
Причините за синдрома на Arnold-Chiari не са напълно установени. Хромозомните аномалии при тази патология, като правило, не могат да бъдат открити.
Патогенеза(какво се случва?) по време на малформация на Arnold-Chiari:
Към днешна дата патогенезата на патологията не е окончателно установена. По всяка вероятност тези патогенетични факторитри:
първият е наследствено причинени вродени остеоневропатии,
второто е травматично увреждане на сфеноетмоидалната и сфеноокципиталната част на кливуса поради родова травма,
третият е хидродинамично въздействие на цереброспиналната течност в стените на централния канал на гръбначния мозък.
Анатомични особености на малформацията на Киари
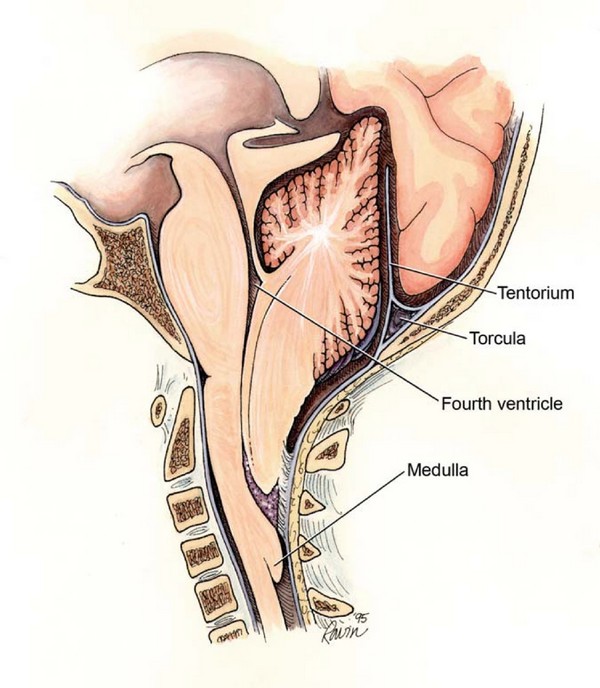
Малкият мозък се намира в задната черепна ямка. (ЗЧЯ)
Сливиците са долната част на малкия мозък. Обикновено те са разположени над foramen magnum. При малформация на Chiari, церебеларните тонзили се намират под foramen magnum, в гръбначния канал.
Foramen magnum е своеобразна граница между черепа и гръбначния стълб, между главния и гръбначния мозък. Над foramen magnum е задната черепна ямка, отдолу е гръбначният канал.
На нивото на foramen magnum долната част на мозъчния ствол (medulla oblongata) преминава в гръбначния мозък. Обикновено цереброспиналната течност (CSF) циркулира свободно в субарахноидалните пространства на главния и гръбначния мозък. На нивото на foramen magnum се свързват субарахноидалните пространства на главния и гръбначния мозък, което осигурява свободното изтичане на цереброспиналната течност от мозъка.
При малформация на Chiari ниско разположените церебеларни тонзили възпрепятстват свободната циркулация на цереброспиналната течност между главния и гръбначния мозък. Сливиците блокират форамен магнум, както тапа запушва гърлото на бутилка. В резултат на това изтичането на цереброспиналната течност се нарушава и се развива хидроцефалия.
Симптоми на малформация на Арнолд-Киари:
Киари идентифицира четири вида аномалии с подробното им представяне. Лекарите все още използват тази класификация днес.
1. Малформация на Arnold-Chiari тип I е слизане на PCF структурите в гръбначния канал под равнината на foramen magnum.
2. При малформация на Arnold-Chiari тип II се появява каудална дислокация на долните части на вермиса, продълговатия мозък и четвъртата камера и често се развива хидроцефалия.
3. Малформация на Arnold-Chiari тип III е рядка и се характеризира с грубо каудално изместване на всички структури на задната черепна ямка.
4. Малформация на Arnold-Chiari тип IV - хипоплазия на малкия мозък без изместването му надолу.
Аномалиите от тип III и IV обикновено са несъвместими с живота.
При приблизително 80% от пациентите малформацията на Arnold-Chiari се комбинира с патология на гръбначния мозък - сирингомиелия, която се характеризира с образуване на кисти в гръбначния мозък, причиняващи прогресивна миелопатия. Тези кисти се образуват при спускане и компресия на структурите на задната черепна ямка шийни прешленигръбначен мозък.
Типично клинична картинаМалформацията на Arnold-Chiari се характеризира със следното симптоми:
Болката в цервикално-тилната област се увеличава при кашлица, кихане,
Намалена болка и температурна чувствителност в горните крайници,
Откажи мускулна силав горните крайници,
Спастичност на горните и долните крайници,
припадък, световъртеж,
Намалена зрителна острота,
В по-напреднали случаи се появяват епизоди на апнея (кратко спиране на дишането), отслабване на фарингеалния рефлекс и неволни бързи движения на очите.
Възможни последствия, усложнения:
1. На фона на нарастващите признаци на вътречерепна хипертония (понякога без нея), прогресивна дисфункция на малкия мозък и компресия на цервикалния гръбначен мозък, се отбелязват парализа на черепните нерви.
2. Понякога малформацията на Arnold-Chiari се комбинира с костни дефекти - окципитализация на атласа и базиларна импресия (фуниевидно вдлъбване на кливуса и краниоспиналната става).
3. Гръбначни аномалии, деформации на ходилата.
Диагностика на малформация на Arnold-Chiari :
Понякога малформацията на Chiari не се проявява по никакъв начин и се открива случайно по време на диагностични процедури.
Понастоящем методът на избор за диагностициране на тази патология е ЯМР на шийката на матката и гръднигръбначен мозък (за изключване на сирингомиелия).
Лечение на малформация на Arnold-Chiari :
Ако единственият симптом на заболяването е с ниска интензивност синдром на болка, за лечение се използва консервативна терапия, която включва различни схемис употребата на нестероидни противовъзпалителни средства и мускулни релаксанти.
Ако няма ефект от консервативната терапия в продължение на 2-3 месеца или пациентът има неврологичен дефицит (изтръпване, слабост в крайниците и др.), е показана операция.
Целта на операцията е– ламинектомия, декомпресивна краниектомия на задната черепна ямка и пластика на дура менинги. При такава операция се увеличава обемът на задната черепна ямка и разширяването на foramen magnum, в резултат на което се прекратява компресията на нервните структури и се нормализира потокът на цереброспиналната течност. В случаите на съпътстваща хидроцефалия се извършва операция на шънт.
В Израел на пациентите се предлага нежно и висококачествено лечение, което след лечение позволява на пациентите да пълноценен живот. Хирургичното лечение на синдрома на Arnold-Chiari се извършва с помощта на ендоскоп, докато травматичният ефект от хирургичното лечение е сведен до минимум. Методът на минимално инвазивно хирургично лечение, проведено в израелски клиники, позволява на пациентите с малформация на Арнолд-Киари впоследствие да водят пълноценен живот дори без лекарствена подкрепа.
Симптоматика на синдрома на Arnold-Chiari
Остео-ставните вродени аномалии са клинично по-малко очевидни сами по себе си и много повече се дължат на тях тежки усложненияна централната и периферната нервна система. Неврологичните прояви са най-трудни за понасяне от пациентите и причиняват неблагоприятния ход на този синдром. Като цяло началото на заболяването е бавно и нехарактерно.
Симптоматика, първоначално много замъглена и дори липсваща за дълго време, често се открива в резултат на намесата на редица решаващи фактори, като травматично увреждане на мозъка или назофарингеални инфекции.
Първата проява, която привлича вниманието към присъствието вродена аномалиянепосредствено след раждането, е наличието на миеломенингоцеле (спина бифида). По-късно се отбелязват други клинични явления, които показват наличието на костно-ставни и неврологични аномалии, а именно:
- спина бифида,
- страничен наклон на главата,
- отклонение на очните ябълки,
- главоболие и периодична или преминаваща болка във врата (особено при по-големи деца и възрастни), появяващи се при движения на главата;
- гадене, повръщане.
При много пациенти, поради блокиране на циркулацията на цереброспиналната течност между 4-ия вентрикул и цистерните на основата на черепа, иволютивното заболяване се развива още през първите месеци от живота. вътрешна хидроцефалия, причинявайки появата на много и разнообразни неврологични феномени. Постепенно черепът на детето се увеличава и се появяват затруднения при хранене, дихателни проблеми, а менингоцелето (когато е налице) може да се разязви.
Прояви на интракраниална хипертония:
- тежки главоболия,
- папиларна конгестия или атрофия на зрителния нерв (късна),
- придружени от прогресиращи нарушения зрителна функция, до пълна слепота.
Церебеларни прояви:
- световъртеж;
- атаксия при ходене и в ортостатично положение;
- дизартрия;
- нарушение на преглъщането
- умишлено треперене,
- нистагъм.
Прояви от страна на периферната нервна система:
- парестезия, анестезия, пареза или спастична парализа,
- подобрени костно-сухожилни рефлекси,
- наличие на рефлекс на Бабински.
Прояви в областта на черепномозъчните нерви:
- едностранна или по-рядко двустранна парализа на лицето;
- парализа окуломоторни нерви, изразяващо се най-често с вътрешен страбизъм или диплопия.
Диагностика на синдрома на Arnold-Chiari.
Лумбалната пункция и биохимичните и бактериологични анализи на цереброспиналната течност в повечето случаи не дават съществени данни. В допълнение, използването лумбална пункцияможе да влоши състоянието на пациента и дори да причини смърт поради внезапно намаляване на налягането и пълно проникване на церебеларните тонзили и продълговатия мозък в гръбначния канал.
Рутинното рентгеново изследване разкрива следните аспекти: малка задна черепна ямка; разширяване на foramen magnum и гръбначния канал; хидроцефалия (голям череп с дехисценция); пръстови отпечатъци върху костната плоча на черепа; сплескване на sella turcica; цервикална, дорзална и лумбална спина бифида.
Допълнителните рентгенови изследвания (газова миелоенцефалография) са противопоказани при деца под 2 години. Въпреки това, при по-големи деца и възрастни те определят индиректни и директни признаци на хидроцефалия, изместване на продълговатия мозък и сливиците на малкия мозък, както и компресия на гръбначния мозък в цервикалната област.
Патологично изследване. От анатомична и топографска гледна точка, синдромът на Арнолд-Киари съществува в четири добре индивидуализирани типа, а именно:
- Първият тип, при който има разтягане и пролапс на маломозъчните тонзили без изместване на продълговатия мозък. Този тип се среща най-често при по-големи деца и възрастни. СЪС клинична точказрение, то може да остане безсимптомно през целия живот и откриването му може да стане напълно случайно.
- Вторият тип, при който долната част на малкия мозък и продълговатия мозък се измества през foramen magnum в гръбначния канал. Този тип е по-чест при кърмачета и се проявява клинично с хидроцефалия и често с наличие на миеломенингоцеле.
- Третият тип, с пълно проникване на малкия мозък в миеломенингоцеле на шийните прешлени.
- Четвъртият тип, при който се отбелязва хипоплазия на малкия мозък, причинена от пълната му херния; червеят на малкия мозък не може да се разграничи, а сливиците и малкомозъчният пласт са едва забележими. Тази форма е изключително рядка.
В допълнение към аномалията на тилната кост и прешлените, засягащи малкия мозък и продълговатия мозък, има и други черепни и гръбначни аномалии, а именно запояване на първия прешлен към тилната кост, движение на гръбначния стълб нагоре в областта на черепа поради хипоплазия на тилната кост; сливане на два или три прешлена (най-често 2-ри и 3-ти шийни прешлени), както се случва при синдрома на Klippel-Feil; цервикална, дорзална или лумбална спина бифида.
Ход и прогноза Синдром на Арнолд-Киари . Протичането на заболяването е бавно. Появата на синдрома при новородено е несъвместима с неговото оцеляване и протичането му бързо води до смърт.
В случай на по-бавно протичане, синдромът на Арнолд-Киари се усложнява от появата на хроничен арахноидит и паренхимни лезии на аксона; това става особено важно, когато се появи нарушение на кръвообращението в нервна тъкан. Обикновено в такива случаи невропсихичните разстройства се забелязват късно и се проявяват под формата на параплегия, тетраплегия и забавено умствено развитие.
Лечение Синдром на Арнолд-Киари . Единствения афективно лечениее хирургическа интервенция. Индикациите за операция не се правят въз основа на рентгенологично определяне на костни аномалии, а само когато последните са придружени от тежки неврологични прояви. Хирургичната интервенция се състои от окципитална краниотомия, съчетана с висока ламинектомия; твърдата черупка се нарязва и оставя отворена. Фиброзните сраствания, често обширни, съществуващи около foramen magnum, се пресичат и отлепват.
В присъствието на тежка хидроцефалия, вентрикулът се отделя с помощта на Холтер или Пуденцова клапа (според класически методоперация за хидроцефалия).
Въпреки че в някои случаи незабавните резултати са благоприятни, всички хирургични интервенции за синдрома на Arnold-Chiari са свързани с голям следоперативен риск поради нарушения в продълговатия мозък, които могат да възникнат в близко бъдеще. постоперативен периоди често причиняват смърт.

