Амиотрофична латерална склероза (ALS), известна още като болестта на Лу Гериг и болестта на Шарко, специфично заболяванекоето води до клетъчна смърт. В Обединеното кралство се използва терминът моторно невронно заболяване (MND), докато в други страни терминът се прилага за 5 вида заболявания, от които ALS е най-често срещаното. Характеризира се с мускулна атрофия, мускулни крампи и постепенно нарастваща слабост поради загуба на мускулна тъкан. Това води до затруднено говорене, преглъщане и в някои случаи дишане. Причината е неизвестна в 90-95% от случаите. Около 5-10% от случаите са наследствени. Около половината от случаите се дължат на един или два специфични гена. Това води до смърт на неврони, които контролират доброволното свиване на мускулите. Диагнозата се основава на признаци и симптоми след тестване, за да се изключат други потенциални състояния. Няма лек за ALS. Лекарство, наречено рилузол, забавя прогресията на ALS и увеличава продължителността на живота с 2-3 месеца. Неинвазивната вентилация може да подобри състоянието и продължителността на живота на пациента. Заболяването обикновено се развива от 60-годишна възраст, в случай на наследствено заболяване– от 50. Средната продължителност на живота от началото на заболяването е 3-4 години. Около 10% от пациентите живеят повече от 10 години. Повечето умират от спиране на дишането. В повечето страни честотата на ALS е неизвестна. В Европа и САЩ заболяването се среща при 2 на 100 000 души годишно. Болестта е описана за първи път от Чарлз Бел през 1824 г. Връзката между симптомите и основните неврологични проблеми е описана от Jean-Martin Charcot през 1869 г., а през 1874 г. той започва да използва самия термин "амиотрофична латерална склероза". В САЩ заболяването стана известно, след като удари известния бейзболист Лу Гериг, а Стивън Хокинг стана известен с научните си постижения. През 2014 г. стартира онлайн кампания за повишаване на осведомеността за ALS. В рамките на тази кампания, наречена ice bucket challenge (буквално – тест с кофа с ледена вода), хората се поляха с кофа с вода и направиха дарения.
Знаци и симптоми
Заболяването причинява слабост и загуба на мускули в цялото тяло поради дегенерация на горните и долните двигателни неврони. Засегнатите индивиди може в крайна сметка да загубят способността си да контролират произволните движения, но пикочният мехур, червата и мускулите за движение на очите функционират до напредналите стадии на заболяването. Когнитивната функция също е запазена при повечето хора, въпреки че някои (около 5%) развиват фронтотемпорална деменция. Много (30-50%) също изпитват незначителни когнитивни промени, които може да останат незабелязани, но са видими при подробни невропсихологични тестове. Рядко ALS се наблюдава при хора с дегенеративно мускулно и костно заболяване (част от синдрома на мултисистемната протеинопатия). Заболяването не засяга сетивните нерви и автономната нервна система, което означава, че повечето хора запазват слуха, зрението, чувствителността, обонянието и вкуса.
Първични симптоми
Ранните симптоми на заболяването са доста общи (слабост и/или мускулна атрофия), така че диагностицирането е трудно. Други симптоми включват затруднено преглъщане, спазми или скованост на засегнатите мускули, мускулна слабост (засягаща ръцете или краката) и/или неясна реч и носни проходи. В първите стадии на заболяването се засяга тази част от тялото, в зависимост от това кои двигателни неврони са увредени първи. Около 75% от хората с болестта първо усещат слабост или атрофия на ръцете или краката. Има неловкост при ходене или бягане, човек може да се спъне или да се препъне или да плъзне крака си малко зад себе си (синдром на падналото стъпало). Ако ръката е засегната, тогава има затруднения в движенията, които изискват ръчна сръчност (например закопчаване на риза, писане на нещо, завъртане на ключ в ключалка). Симптомите могат да персистират само в единия крайник за дълъг период от време или през цялото заболяване; симптомът е известен като амиотрофия на единия крайник. Около 25% от всички заболявания започват като прогресиращ булбарен синдром. Първичните симптоми в този случай се проявяват в затруднено говорене или преглъщане. Речта става объркана, по-тиха и назална. Затрудненото преглъщане е придружено от загуба на подвижност на езика. При малка част от хората първо се засягат междуребрените мускули, които допринасят за дихателния процес. Малък процент от хората имат фронтотемпорална деменция, но с напредването на заболяването се появяват по-типични симптоми на ALS. С течение на времето хората изпитват затруднения при движение, преглъщане (дисфагия), говорене и формиране на думи (дизартрия). Дисфункцията на горния моторен неврон се проявява чрез мускулна твърдост (спастичност) и повишена рефлекторна дейност(хиперрефлексия), както и хиперактивен рефлекс на повръщане. Рефлексът на Бабински също показва увреждане на горния двигателен неврон. Симптомите на увреждане на долния двигателен неврон са мускулна слабост и атрофия, мускулни крампи, които могат да се видят под кожата, но малките крампи не са диагностициран симптом, те се наблюдават по-късно или съпътстват слабост и атрофия. Около 15-45% от хората изпитват афективна лабилност, неврологично разстройство, известно като емоционална нестабилност, което се състои от пристъпи на неконтролируем смях, плач, постоянна усмивка, което е свързано с дегенерация на горните моторни неврони на луковицата, изразяваща се в прекомерно изразяване на емоции . За да бъде диагностициран ALS, човек трябва да има симптоми на увреждане на горните и долните неврони, които не са присъщи на други заболявания.
Развитие на болестта
Въпреки че редът и скоростта на развитие на симптомите варират при отделните хора, повечето хора не могат да ходят или да използват ръцете си. Те също така губят способността да говорят и да преглъщат храна, така че най-често се използва BiPAP неинвазивна вентилация (BiPAP). Скоростта на прогресиране на заболяването се оценява с помощта на скалата ALSFRS-R, която представлява въпросник или клинично интервю със скала от 48 (нормално функциониране) до 0 (тежка форма). Въпреки че степента на вариация е висока и малък брой хора имат ниска степен на заболяване, средно пациентите губят 0,9 точки на месец. Проучване, базирано на клинични прегледи, показва 20% промяна в ALSFRS-R като клинично значима. Независимо коя част от тялото е засегната първа, болестта в своето развитие се разпространява и в други части на тялото. Симптомите на крайниците обикновено се разпространяват в противоположни крайници, а не в нова част на тялото, докато булбарното начало на заболяването засяга първо ръцете и след това краката. Скоростта на прогресиране на заболяването е по-ниска при пациенти на възраст под 40 години в началото, при пациенти с ниски нива на затлъстяване, при индивиди със заболяване на единия крайник и при тези с първични симптоми на заболяване на горния моторен неврон. Обратно, скоростта на прогресиране на заболяването е по-висока и прогнозата е по-лоша при хора с булбарна, респираторна и фронтотемпорална деменция. Алелният вариант CX3CR1 оказва влияние върху прогресията на заболяването и продължителността на живота.
Късни етапи
Въпреки че асистираната вентилация може да облекчи проблемите с дишането и да увеличи продължителността на живота, тя не забавя прогресията на ALS. Повечето хора с ALS умират от спиране на дишането в рамките на 3-5 години от началото на симптомите. Средната продължителност на живота от началото на заболяването до смъртта е 39 месеца, а само 4% от хората живеят повече от 10 години. Китаристът Джейсън Бекер живее с болестта от 1989 г., а физикът Стивън Хокинг живее с болестта повече от 50 години, но всички тези случаи са уникални. Затрудненото дъвчене и преглъщане затруднява храненето и увеличава риска от задавяне и поглъщане на храна в белите дробове. В по-късните стадии на заболяването може да се развие аспирационна пневмония и поддържането на теглото може да се превърне в сериозен проблем, което може да изисква сонда за хранене. С отслабване на диафрагмата и междуребрените мускули гръден кош, които подпомагат дишането, белодробната функция, или по-скоро жизненият капацитет и инспираторното налягане, намаляват. При респираторната форма на заболяването това може да се случи и преди отслабването на мускулите на крайниците. Повечето пациенти с ALS умират от спиране на дишането или пневмония. На финални етапизаболяването може да бъде засегнато окуломоторния нерв, който контролира движението, подобно на мускулите на очната ябълка. Движението на очите се запазва до последните етапи поради мускулните различия очна ябълкасъс скелетните мускули, които са засегнати първи от болестта. В по-късните стадии на заболяването състоянието на пациента може да наподобява синдром на блокиране.
движение на очите
Хората с ALS може да имат затруднения при извършването на бързи доброволни движения на очите. Скоростта на движение на очите се забавя. Има и спазъм на конвергенция на очите. Тестването на вестибуло-очния рефлекс може да помогне за идентифицирането на тези проблеми. Електроокулографията (ЕОГ) измерва потенциала на покой на ретината. При хора с ALS, EOG демонстрира промени, които засягат прогресията на заболяването и също така предоставя данни за клинична оценка на прогресията на заболяването, което засяга окуломоторната активност. В допълнение, EOG ви позволява да идентифицирате проблеми с очите на ранен етап. Окуломоторните мускули са различни от другите скелетни мускули. Окуломоторният мускул е уникален с това, че непрекъснато се модифицира през целия живот и поддържа броя на сателитните клетки по време на стареенето. В окуломоторните мускули има много повече миогенни прогениторни клетки, отколкото в скелетните мускули на крайниците.
причини
Генетика
В 5-10% от случаите заболяването се унаследява директно от родителите. Около 20% от фамилните случаи (или 2% от всички случаи) са свързани с мутация на хромозома 21, която кодира супероксид дисмутаза. Има над сто форми на мутация. IN Северна Американай-често срещаният мутантен ген е мутантен SOD1 ген; характеризира се с невероятно висок процент на прогресиране от момента на заболяването до момента на смъртта. Най-често срещаният мутант в скандинавските страни е D90A-SOD1, с по-бавна скорост на прогресия от типичния ALS, а хората с тази форма на заболяването живеят средно 11 години. През 2011 г. в C9orf72 беше открита генетична аномалия, известна като хексануклеотидно повторение, тази аномалия е свързана с ALS и фронтотемпорална деменция и представлява до 6% от всички случаи на ALS сред белите европейци. Този ген присъства и при хора от филипински произход. Генът UBQLN2 е отговорен за производството на протеина убиквилин 2 в клетката, който е член на семейството на убиквилин и контролира разграждането на убиквитинирани протеини. Мутациите в UBQLN2 предотвратяват разграждането на протеина, което води до невродегенерация и причинява (предимно наследствена) Х-свързана ALS и ALS/деменция.
SOD1
През 1993 г. учените откриха, че мутация в гена (SOD1), който произвежда ензима Cu-Zn супероксид дисмутаза (SOD1), е свързана с 20% от случаите на наследствена ALS. Този ензим е доста мощен антиоксидант, който предпазва тялото от увреждане, причинено от супероксид, токсичен свободен радикал, генериран в митохондриите. Свободните радикали са силно реактивни молекули, произведени от клетките по време на метаболизма. Свободните радикали могат да се натрупват и да причинят увреждане на ДНК и протеините в клетката. В момента над 110 различни мутации в SOD1 са свързани с ALS, някои от които (като H46R) имат много дълга клинична история, докато други, като A4V, са изключително агресивни. Когато защитата срещу оксидативния стрес е отслабена, се активира клетъчна смърт (апоптоза). Дефект в SOD1 може да доведе до загуба или придобиване на функционалност. Загубата на SOD1 функция може да доведе до натрупване на свободни радикали. Придобиването на SOD1 функции може да бъде токсично. В резултат на проучвания, включващи трансгенни мишки, бяха предложени няколко теории за ролята на SOD1 в наследствената ALS. Мишките без ген SOD1 не развиват ALS, въпреки че изпитват ускорена свързана с възрастта мускулна атрофия (саркопения) и намалена продължителност на живота. Това означава, че токсичните свойства на мутацията SOD1 са резултат от усилване на функцията, а не загуба. В допълнение, установено е, че натрупването на протеин е патологична характеристика на наследствената и спорадична ALS (протеинопатия). Любопитно е, че при мишки с мутирал SOD1 (най-често мутация на G93A), натрупването (неправилно нагънат протеин) на мутиралия SOD1 се открива само в засегнатите тъкани, като по-голямо натрупване се наблюдава по време на моторна невронална дегенерация. Учените вярват, че натрупването на мутирал SOD1 играе важна роля в нарушаването на клетъчните функции чрез увреждане на митохондриите, протеазомата, шапероните и други протеини. Ако се потвърди, всяка от тези аномалии може да послужи като доказателство, че подобни натрупвания водят до токсичността на мутиралия мутирал SOD1. Критиците посочват, че при хората мутациите на SOD1 причиняват само 2% от всички случаи на заболяване и причинно-следствените механизми могат да се различават от тези, отговорни за спорадичната форма на заболяването. В момента мишките ALS-SOD1 остават най-добрият модел на заболяването в предклиничните проучвания, но се надяваме, че ще бъде разработен нов модел. Налична е онлайн база данни, която е научна общност и платформа с актуална информация за ALS за широката общественост. Сайтът се нарича ALSOD, първоначално е създаден за публикации за гена SOD1 през 1999 г., в момента има повече от 40 гена, свързани с ALS, разположени на сайта.
Други фактори
В случай, че заболяването не е наследствено, тоест в 90% от случаите, причините за заболяването са неизвестни. Възможни причинимакар и ненадеждни, са травма на главата, военна служба, честа употреба на наркотици и участие в контактни спортове. Изследванията се фокусират и върху ролята на глутамата в дегенерацията на моторните неврони. Глутаматът е един от невротрансмитерите в мозъка. Учените са установили, че хората с ALS, в сравнение със здравите хора, имат повече високо нивоглутамат в кръвта и цереброспиналната течност. Riluzole в момента е единственото лекарство, одобрено в Съединените щати за лечение на ALS и ефекти върху глутаматните транспортери. Лекарството има слаб ефект върху увеличаването на продължителността на живота, което предполага, че излишъкът на глутамат не е единствената причина за заболяването. Някои изследвания предполагат връзка между спорадични ALS (особено при спортисти) и диета, богата на аминокиселини с разклонена верига (популярна добавка сред спортистите), които причиняват клетъчна възбудимост, подобна на тази при хора с ALS. Клетъчната възбудимост води до повишена абсорбция на калций от клетката, което води до смъртта на невронните клетки. Някои доказателства предполагат, че всепроникващото разрушаване на образуването на протеинова структура на супероксид дисмутаза 1 (SOD1) се случва по същия начин, както при прионите. Включването на β-метиламино-1-аланин (BMAA) също се предполага, че води до друго подобно на прион разпространение на образуването на нарушена протеинова структура. Друг често срещан фактор, свързан с ALS, е лезията двигателна системав области като фронтотемпоралните дялове. Поражението в тази област е знак ранни нарушения, което може да се използва за прогнозиране на загубата двигателна функция. Механизмите на ALS се проявяват много преди да се появят първите признаци и симптоми. Преди мускулната атрофия да стане очевидна, около една трета от моторните неврони трябва да умрат. Изследвани са много други потенциални рискови фактори - излагане на химикали, електромагнитни полета, физическо нараняване и токов удар - но не са направени последователни заключения.
Патофизиология
Отличителна черта на ALS е смъртта на горните и долните двигателни неврони в проекционната зона на мозъчната кора, мозъчния ствол и гръбначния мозък. Преди смъртта си моторните неврони развиват богати на протеини включвания в клетъчното тяло и аксоните. Отчасти това може да се дължи на разграждането на протеина. Тези включвания често съдържат убиквитин и често включват един от ALS протеините: SOD1, TAR ДНК свързващ протеин (TDP-43 или TARDBP) или FUS RNA свързващ протеин.
Скелетни двигателни единици
Външни мускули на очната ябълка и скелетни мускулипоказват различни характеристики. По-долу са изброени характеристики, които отличават мускулите на очната ябълка от скелетните мускули.
Едно нервно влакно се свързва само с едно мускулно влакно
Без рефлекс на разтягане, въпреки голям броймускулни вретена
Без циклично инхибиране
Липса на бързи/бавни мускули
Всички моторни неврони на окото участват във всички видове движения на очите.
Наблюдават се и разлики между здрави и засегнати мускули на очната ябълка. Мускулите на очната ябълка на починалите донори запазват своята цитоархитектоника в сравнение с мускулите на крайниците. Здравите мускули на очната ябълка се състоят от централен слой (GL) пред очната ябълка и тънък орбитален слой (OL) пред орбитата. Окуломоторните мускули, засегнати от ALS, запазват позицията на GL и OL. Окуломоторните мускули запазват мозъчния невротрофичен фактор (BDNF) и глиалния невротрофичен фактор, които също са запазени в мускулите, засегнати от ALS. Ламининът е структурен протеин, разположен в невромускулната връзка (NMJ). Lnα4 е изоформа на ламинин, който е отличителен белегневромускулна връзка на окуломоторните мускули. Хората с ALS запазват експресията на Lnα4 в нервно-мускулната окуломоторна връзка, но тази експресия отсъства в мускулите на крайниците на същите хора. Поддържането на експресията на ламинин може да играе важна роля в поддържането на целостта на окуломоторните мускули при хора с ALS. Хората със спорадичен ALS (sALS) имат повишено нивовътреклетъчен калций, който предизвиква повишено освобождаване на невротрансмитери. Пасивният транспорт на серум от хора със sALS увеличава спонтанното освобождаване на медиатори в цереброспиналната течност. Окуломоторните мускули са устойчиви на промени във физиологичното състояние. Има обаче различни видовевъздействието на болестта. Окуломоторните мускули, засегнати от ALS, имат по-голяма вариация в размера на влакната, отколкото здравите контролни мускули. Групирани и разпръснати атрофични и хипертрофични влакна са открити в окуломоторните мускули, но увреждането на тези мускули е значително по-ниско, отколкото в мускулите на крайниците на същите донори. Също така има увеличение на съединителната тъкан и увеличаване на мастната тъкан в окуломоторните мускули, за да се компенсира загубата на влакна и атрофията. Пациентите с ALS също имат офталмоплегия, загуба на неврони около и в ядрата на двигателните мускули на очната ябълка. В допълнение, съдържанието на тежката верига на миозина във влакната на окуломоторните мускули се променя, нормалната експресия на тежката верига на бавния миозин в GL е нарушена и тежката верига на ембрионалния миозин отсъства в OL. Промените в тежката верига на бавния миозин и тежката верига на ембрионалния миозин са единствените промени в окуломоторните мускули. Тъй като окуломоторният мускул е силно инервиран, всяка денервация се компенсира от съседни аскони, които остават функционални.
лактат и цинамат
Млечна киселина - краен продуктгликолиза, която причинява мускулна умора. Ензимът лактат дехидрогеназа работи двупосочно и може да окисли лактат до пируват, така че може да се използва в цикъла на Кребс. В окуломоторните мускули лактатът подпомага мускулната контракция по време на повишена активност. Смята се, че окуломоторните мускули висока активностлактат дехидрогеназите са резистентни към ALS. Цинаматът е блокер на транспорта на лактат. Цинаматът може да причини умора на глозо-моторните мускули, намалявайки мускулната издръжливост и остатъчното усилие. Въпреки това, цинаматът няма ефект върху дългите мускули на разгъвачите на пръстите на краката. Заместването на глюкозата с екзогенен лактат увеличава умората на мускулите extensor digitorum longus, но няма ефект върху глозомоторния мускул. Умората на окуломоторните мускули се наблюдава само когато комбинацията от екзогенен лактат и цинамат замества глюкозата.
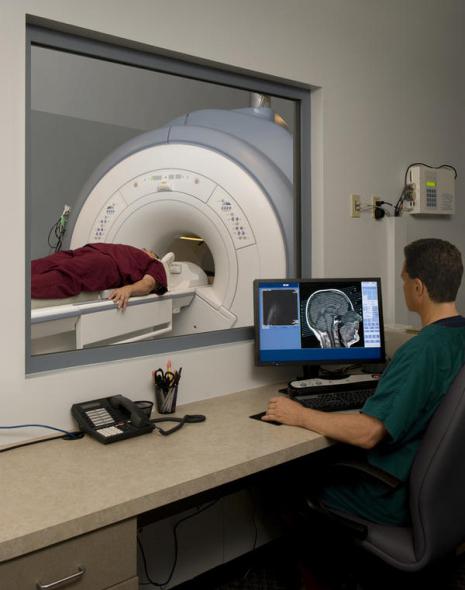
Диагностика
Никой анализ не може да диагностицира точно ALS, въпреки че наличието на признаци, които показват смъртта на горните и долните моторни неврони, е съществен признак. Диагнозата ALS се поставя основно въз основа на наблюдения на лекар и серия от тестове, които изключват други заболявания. Лекарят снема пълна медицинска история на пациента и редовно извършва неврологични прегледи, за да оцени прогресията на симптоми като слабост, мускулна атрофия, хиперрефлексия и мускулна спастичност. Могат да се направят допълнителни тестове, за да се изключи възможността за други състояния, тъй като много от симптомите на ALS могат да се появят и при други състояния, които могат да бъдат лекувани. Един такъв тест е електромиографията (ЕМГ), която открива електрическата активност в мускулите. Някои ЕМГ находки могат да подкрепят диагнозата ALS. Друг общ тест е тестът за скорост на нервната проводимост (NVR). Определено отклонение, открито в резултат на теста, може да показва, че пациентът има форма на периферна невропатия (увреждане на периферни нерви) или миопатия (мускулно заболяване), а не ALS. Магнитно-резонансното изображение може да открие аномалии, които причиняват симптоми на ALS, като подуване гръбначен мозък, множествена склероза, цервикална дискова херния, сирингомиелия или цервикална спондилоза. Въз основа на симптомите и резултатите от изследванията, Вашият лекар може да назначи изследвания на кръв и урина или други лабораторни изследвания, за да изключи други възможни заболявания. В някои случаи, ако лекарят подозира, че пациентът има миопатия, а не ALS, той може също да нареди мускулна биопсия. Вирусни заболявания като вирус на човешка имунна недостатъчност (HIV), вирус Т-клетъчна левкемиячовешки (HTLS), лаймска болест, сифилис и енцефалит, пренасян от кърлежиможе да причини симптоми, подобни на тези на ALS. Неврологични заболяваниякато множествена склероза, синдром след полиомиелит, мултифокална моторна невропатия, синдром на Guillain-Barré и спинална мускулна атрофия също могат да имат подобни на ALS симптоми. Важно е правилно да се диагностицира заболяването, тъй като симптомите на ALS могат лесно да бъдат объркани със симптомите на редица други заболявания. Поради това е необходим преглед от квалифициран невролог, за да се изключат други заболявания. В повечето случаи ALS се диагностицира лесно, като погрешната диагноза представлява по-малко от 10% от случаите. Проведено е проучване с протокол на изследване и редовни прегледи, в което са участвали 190 пациенти, отговарящи на критериите за MND/ALS. Диагнозата на 30 пациенти (16%) се промени драстично по време на периода на клинично проследяване. В същото проучване трима пациенти са имали фалшиво отрицателна диагноза миастения гравис (TM, автоимунно заболяване). TM има същите симптоми като ALS и няколко други неврологични аномалии, което води до забавяне на диагнозата и лечението. TM е лечимо, но ALS не е. Миастеничният синдром, иначе синдромът на Lambert-Eaton, може да имитира ALS и ранни симптомиподобни на симптомите на ТМ.
Лечение
Терапията е необходима за пациенти с ALS за облекчаване на симптомите и увеличаване на продължителността на живота. Мултидисциплинарният медицински екип, който работи с пациенти с ALS, вярва, че поддържащите грижи са от съществено значение, за да могат пациентите да останат активни и комфортни.
Медицински препарати
Riluzole (Rilutek) - леко увеличава продължителността на живота на пациентите. Удължава продължителността на живота с няколко месеца и има по-голямо въздействие върху пациенти с булбарна ALS. Лекарството също така ви позволява да забавите използването на вентилация. Пациентите, които приемат лекарството, трябва да преминат изследване на черния дроб (10% от хората изпитват увреждане на черния дроб, когато се приемат). Лекарството е одобрено от Министерството на здравеопазването на САЩ и се препоръчва за употреба. Национален институтклинична квалификация. Riluzole не възстановява увреждането, което вече е нанесено на моторните неврони. Други лекарства могат да се използват за намаляване на умората, облекчаване на мускулни крампи, контрол на спастичността и намаляване на повишено слюноотделянеи храчки. Редица лекарства също могат да облекчат болката, депресията, да подобрят съня, да намалят дисфагията и запека. Баклофен и диазепам се предписват за контрол на спастичността на ALS. Трихексифенидил или амитриптилин могат да бъдат предписани, ако пациентите имат проблеми с преглъщането на слюнка.
Подкрепа на дишането
Когато мускулите, участващи в дишането, са отслабени, може да се използва вентилация (вентилация с положително налягане, двустепенно положително налягане на дихателните пътища (BiPAP) или IVL бифазна вентилация (BCV)) за подпомагане на дишането. Такива устройства изкуствено принуждават белите дробове да функционират с помощта на външни устройства, разположени на лицето и тялото. Когато мускулите вече не могат да поддържат нивата на кислород и въглероден двуокис, тези устройства могат да се използват постоянно. BCV има ясното предимство, че устройството може да изчисти секрети с високочестотни вибрации, които се появяват след няколко издишвания. Пациентите могат също така да обмислят използването на механична вентилация (респиратори), при която устройството надува и издухва белите дробове. За ефективно използваненеобходима е тръба, която трябва да премине от носа или устата през трахеята. Дългосрочната употреба на такова устройство може да изисква операция, трахеотомия, по време на която тръба се вкарва директно в трахеята на човека през отвор на шията. Пациентите и техните семейства трябва да имат предвид няколко фактора, когато решават кога да използват едно от устройствата, описани по-горе, и дали изобщо да го използват. Вентилаторите се различават по въздействието си върху качеството на живот и по цена. Докато вентилацията може да помогне за облекчаване на проблемите с дишането и по този начин да удължи живота, тя не влияе върху прогресията на ALS. Преди да решат кое устройство да изберат, пациентите трябва да бъдат напълно информирани за тях и влиянието им върху живота без движение. Някои хора с продължителна трахеотомия, вентилация с положително налягане със специални машини или тръби, могат да говорят, ако мускулите не са засегнати устната кухина(но във всеки случай, с прогресирането на заболяването, речта ще бъде загубена). Други пациенти могат да използват говорна клапа (като говорна клапа на Passy-Mure) под ръководството на логопед. За поддържане на дишането, първо през нощта и след това през деня, се използват външни вентилатори, които работят в режим на вентилация BiPAP. Използването на BiPAP е временна мярка. Много преди BiPAP вече да не е ефективен, пациентите трябва да обмислят трахеотомия и дългосрочна механична вентилация. На този етап някои пациенти избират хосписно лечение.
Терапия
Физиотерапията играе важна роля в процеса на рехабилитация, има благоприятен ефект върху пациентите с ALS, като позволява да се забави загубата на сила, да се поддържа издръжливостта, да се намали болката, да се предотвратят усложненията и да се осигури функционална независимост. Рехабилитационната терапия и специалното оборудване също спомагат за осигуряване на независимост и безопасност на пациентите в хода на ALS. Леките аеробни упражнения, като ходене, плуване и колоездене, укрепват незасегнатите мускули, подобряват сърдечно-съдовото здраве и помагат на пациентите да се справят с умората и депресията. Редуването на упражненията за движение и разтягане предотвратява мускулни крампи и мускулни контракции. Терапевтите трябва да гарантират, че мускулите са натоварени, без да им позволяват да претоварват. Рампи, скоби, проходилки, оборудване за баня, инвалидни количкикоето помага на пациентите да останат мобилни. Терапевтите могат да препоръчат оборудване или устройства, които да помогнат на пациентите да останат възможно най-безопасни и нормални. Пациентите, които имат затруднения при говорене, могат да работят със специалисти по говорни увреждания, за да им помогнат да научат пациентите на техники, като например как да говорят по-силно и по-ясно. С напредването на заболяването специалистите могат да препоръчат използването на устройства за подобряване на речта или алтернативни начиникомуникация, като високоговорители, устройства за генериране на реч и/или табла с азбука, комуникация чрез сигнали да/не.
Хранене
Пациентите и лицата, които се грижат за тях, могат да получат необходимата информацияот диетолози за това как да планирате диетата си и да ядете малки хранения през деня, които ще ви осигурят достатъчно калории, фибри и течности, както и информация за това какви храни да избягвате, за да избегнете проблема със затрудненото преглъщане. Пациентите могат да използват устройства за засмукване, за да отстранят излишната течност и слюнка, като по този начин предотвратяват задавяне. Терапевтите могат да помогнат с препоръки за самостоятелно хранене. Специалистите по говорни увреждания могат да ви помогнат да изберете продукти, които са най-подходящи за вашите способности. Когато пациентът вече не може да получава хранителни веществаот храна, лекарите може да препоръчат използването на сонда за хранене. Използването на сонда за хранене също предотвратява риска от задушаване и пневмония, които могат да възникнат от вдишване на течност в белите дробове. Слушалката не причинява никакви болкаи позволява на пациентите да се хранят сами, ако желаят. Изследователите заявяват, че „пациентите с ALS имат хроничен дефицит на енергиен прием“ и намален апетит. Изследванията върху животни и хора потвърждават, че пациентите трябва да приемат възможно най-много калории и никога да не намаляват приема на калории. Към 2012 г. няма точни данни относно лечението за отслабване.
Палиативна грижа
Социалните работници, болногледачите и медицинските сестри в хосписа помагат на пациенти с ALS, техните семейства и болногледачи да се справят с медицински, емоционални и финансови затруднения, особено в напредналите стадии на заболяването. Социалните работници оказват подкрепа при получаване на финансова помощ, написване на пълномощно и завещание и помагат да се намери подкрепа за семейства и лица, които се грижат за тях. Грижачите не само осигуряват медицински грижи, но също така да научи членовете на семейството на пациента как правилно да използват респиратори, да хранят и движат пациента по такъв начин, че да избегнат болезнени кожни проблеми и стягане. Медицинските сестри в хосписа работят с лекари, за да осигурят подходящи грижи и да помогнат при други въпроси, свързани с подобряването на качеството на живот на пациентите, които желаят да останат у дома. Служителите в хосписа също така консултират пациентите и техните семейства по всички въпроси, свързани с крайната фаза на заболяването.
Епидемиология
В повечето страни честотата на ALS е неизвестна. В Европа заболяването засяга около 2,2 души на 100 000 души годишно. В САЩ всяка година се диагностицират 5600 души, докато 30 000 американци вече имат заболяването. Отстрани амиотрофична склерозадвама от 100 000 души умират всяка година. ALS се разглежда рядко заболяване, но най-честата от всички болести на моторните неврони, която засяга хора от всякаква раса и етнически произход. ALS се развива при 1-2 души на 100 000 годишно. ALS засяга до 30 000 американци. Съобщава се, че заболяването засяга 1,2–4,0 на всеки 100 000 кавказци, като по-малък брой се наблюдава при други етнически групи. Филипинците са на второ място по разпространение на болестта (1,1-2,8 на всеки 100 000). Има съобщения за няколко „групи“, включително трима играчи на американски футбол от San Francisco 49ers, над 50 играчи на футболна асоциация в Италия, трима футболни приятели в южна Англия и семейни случаи (съпруг и съпруга) в южна Франция. Повечето изследователи твърдят, че ALS възниква поради комбинация от наследствени и фактори на околната среда, въпреки че последното не е потвърдено, за разлика от повишен риск от заболяването с възрастта.
История
Болестта е описана за първи път през 1824 г. от Чарлз Бел. В Съединените щати болестта стана известна, след като удари известния бейзболист Лу Гериг, а след това и след кампания, наречена предизвикателството с ледена кофа през 2014 г. Английският учен Август Уолър описа външен вид нервни влакнапрез 1850 г. През 1869 г. връзката между симптомите и подлежащите неврологични проблеми е описана от Жан-Мартен Шарко, който въвежда концепцията за амиотрофична латерална склероза в работата си през 1874 г. През 1881 г. статията е преведена на английски езики публикуван в три тома на Лекции по болести нервна система". ALS става известен в Съединените щати през 1939 г., след като болестта поразява бейзболната легенда Лу Гериг, който умира две години по-късно. През 50-те години на миналия век епидемия от АЛС възниква сред народа Чаморо в Гуам. До 1991 г. изследователите вече свързват хромозома 21 с наследствената форма на ALS (HALS). През 1993 г. беше открито, че генът SOD1 на хромозома 21 играе важна роля в редица случаи на наследствена форма на заболяването. През 1996 г. рилузол е одобрен от Министерството на здравеопазването на САЩ за лечение на ALS. През 1998 г. критерият El Escorial е установен като стандарт за класифициране на пациенти с ALS в клинични изпитвания. На следващата година беше публикувана скалата за функционалност на ALS, която се превърна в стандарт за оценка на заболяването в клинични проучвания. През 2011 г. беше установено, че множеството повторения на C9ORF72 са основната причина за ALS и фронтотемпорална деменция.
Етимология
Терминът "амиотрофичен" идва от гръцката дума amyotrophia: a- означава "не", myo означава "мускул" и trophia означава "хранене"; по този начин амиотрофията означава "липса на мускулно хранене", което точно описва характерна особеностзаболяване, мускулна атрофия. „Странично“ се отнася до областта на човешкия гръбначен мозък, където се намират засегнатите нервни клетки. Дегенерацията в тази област води до уплътняване, склероза.
Обществена подкрепа и културна референция
През август 2014 г. в интернет се проведе акция в подкрепа на хората с ALS, наречена ALS Ice Bucket Challenge. Протестиращият трябваше да напълни кофа с вода с лед, след което да назове човека, който ги е предизвикал, както и тримата души, които е предизвикал. Тогава участникът събори върху себе си кофа с вода и лед. Но беше възможно да се участва в акцията по друг начин. Член може да дари минимум 10 щатски долара (или еквивалент в друга валута) за изследване на ALS в Обединеното кралство. Този, който не иска да се мокри студена водатрябва да дари минимум $100 за изследване на ALS. Към 25 август акцията е събрала $79,7 млн., докато през 2013 г. са събрани едва $2,5 млн. В акцията са участвали много знаменитости. ALS е в центъра на филма от 2014 г. „Ти не си ти“ с участието на Хилари Суонк, Емили Росъм и Джош Дюамел.
Проучване
По света се провеждат клинични изпитвания; списък на клиничните изпитвания в САЩ може да бъде намерен на ClinicalTrials.gov. Най-голямото генетично изследване, наречено MinE project, все още продължава. Проектът се финансира чрез публично набиране на средства и включва много държави. Фаза-II на проучването беше завършена, а фаза-IIb все още продължава под името „BENEFIT-ALS“. Резултатите от първото проучване са достъпни тук Настоящото проучване е международно, плацебо-контролирано проучване, включващо 680 пациенти. Това го прави най-мащабното проучване досега. В момента се провежда фаза II проучване на антитялото озанезумаб. Това е голямо проучване, спонсорирано от британската компания GlaxoSmithKline. Болница Хадаас в Израел преминава във фаза II клинично изпитванепроведено от BrainStorm Cell Therapeutics, работата е насочена към стабилизиране на параметрите на функционалната скала на ALS. Стволовите клетки се отстраняват по време на теста костен мозъкчовешки и се диференцират в свободното пространство на клетката, което активира невротропни фактори. Клетките се инжектират обратно в същия пациент чрез интратекални и интрамускулни инжекции. Планира се втората част от фаза II да се проведе в няколко американски институции, включително клиниката Mayo.
Болестта амиотрофична латерална склероза (ALS) се среща при трима на сто хиляди души. Въпреки днешния напредък на медицината, смъртността от тази патология е 100%. Има обаче случаи, когато пациентите не умират с течение на времето, състоянието им се стабилизира. Ярък примере известният китарист Джейсън Бекер. Той се бори активно с това заболяване повече от 20 години.
Какво е BAS?
При това заболяване има последователна смърт на моторните неврони на гръбначния мозък и отделни части на мозъка, които са отговорни за произволните движения. С течение на времето мускулите при хората с тази диагноза атрофират, тъй като са постоянно бездействащи. Болестта се проявява под формата на парализа на крайниците, мускулите на тялото и лицето.
Амиотрофичната латерална склероза се нарича амиотрофична латерална склероза само защото невроните, които провеждат импулси към всички мускули, са разположени от двете страни на гръбначния мозък. Последният стадий на заболяването се диагностицира, когато патологичният процес достигне дихателните пътища. Смъртта настъпва поради мускулна атрофия или инфекция. В някои случаи дихателните мускули са засегнати преди крайниците. Човек умира много бързо, без да изпита всички трудности на живота с парализа.
В много европейски страни амиотрофичната латерална склероза е известна като болестта на Лу Гериг. Този известен бейзболен играч от Америка е диагностициран още през 1939 г. Само за няколко години той напълно загуби контрол над тялото си, мускулите му бяха изтощени, а самият спортист стана инвалид. Лу Гериг почина през 1941 г.
Рискови фактори
През 1865 г. Шарко (френски невролог) за първи път описва това заболяване. Днес от него страдат не повече от петима от сто хиляди души по света. Възрастта на пациентите с тази диагноза варира от 20 до 80 години. Представителите на по-силния пол са по-податливи на това заболяване.
В 10% от случаите заболяването ALS е наследствено. Учените са идентифицирали около 15 гена, чиято мутация в различни степенисе проявява при хора с тази патология. 
Останалите 90% от случаите са спорадични, т.е. не са свързани с наследствеността. Експертите не могат да назоват конкретни причиникоето води до развитие на болестта. Предполага се, че някои фактори все още могат да увеличат риска от заболяването, а именно:
- Пушенето.
- Работа в опасна индустрия.
- Служба в армията (на учените е трудно да обяснят това явление).
- Яденето на храни, които са отгледани с пестициди.
Основните причини за заболяването
Тежък патологичен процес може да започне напълно различни факторис които се сблъскваме ежедневно Истински живот. Защо възниква ALS? Причините може да са следните:
- Интоксикация на организма с тежки метали.
- Инфекциозни заболявания.
- Дефицит на определени витамини.
- Електрическо нараняване.
- Бременност
- Злокачествени новообразувания.
- Хирургични интервенции (отстраняване на част от стомаха).
Форми на заболяването
Цервико-торакалната форма се характеризира с разпространението на патологичния процес в областта на лопатките, ръцете и всичко. раменния пояс. Постепенно става трудно човек да извършва обичайни движения (например закопчаване на копчета), върху които не е трябвало да се концентрира преди заболяването. Когато ръцете спрат да се "подчиняват", настъпва пълна мускулна атрофия. 
Лумбосакралната форма се характеризира с поражение долни крайницикато ръце. Постепенно се развива слабост на мускулите в тази област, появяват се потрепвания и конвулсии. Пациентите започват да изпитват трудности при ходене, постоянно се спъват.
Булбарната форма е една от най-тежките прояви на заболяването. Много рядко пациентите успяват да оцелеят повече от четири години от началото на първичните симптоми. Признаците на заболяването ALS започват с проблеми с говора и неконтролируеми изражения на лицето. Пациентите имат затруднено преглъщане, което се превръща в пълна невъзможност за самостоятелно хранене. Патологичният процес, обхващащ цялото човешко тяло, влияе негативно на работата на дихателните и сърдечно-съдови системи. Ето защо пациентите с тази форма умират, преди да се развие парализа.
Церебралната форма се характеризира с едновременно участие в патологичния процес както на горните, така и на долните крайници. В допълнение, пациентите могат да плачат или да се смеят без причина. По тежест церебрална формане отстъпва на луковицата, така че смъртта от нея идва също толкова бързо.
Клинична картина
Според някои данни дори на предклиничния етап приблизително 80% от моторните неврони умират. Цялата им работа се поема от съседни клетки. Те постепенно увеличават броя на крайните разклонения и така нареченият йонен код започва да се превежда към голям брой мускули. Поради създаденото претоварване тези неврони също умират. Така започва амиотрофичната латерална склероза. Симптомите на заболяването не се появяват веднага след смъртта на моторните неврони.
Може да отнеме 5-7 месеца, преди човек да обърне внимание външни променина вашето тяло. Пациентите, като правило, имат намаляване на телесното тегло и мускулна слабост, има трудности при изпълнението на ежедневните дейности. Става трудно да се движите нормално, да носите предмети в ръцете си, да дишате, преглъщате и говорите. Появяват се конвулсии и потрепвания. Такива симптоми са характерни за много заболявания, което значително усложнява диагностиката на ALS в ранните етапи на развитие.
При тази патология системите никога не са засегнати вътрешни органи(бъбреци, черен дроб, сърце), мускули, отговорни за чревната подвижност.
Болестта ALS има прогресиращ характер и с течение на времето обхваща все повече и повече области на тялото. Човек постепенно губи способността да се движи без затруднения, поради нарушение гълтателни рефлексипостоянно има храна Въздушни пътищакоето води до прекъсване на дишането. В последните етапи жизнената активност се поддържа само благодарение на изкуствено хранене и вентилатор. 
Диагностика
Диагностицирането на заболяването е изключително трудно. Цялата работа е в това начални етапи ALS заболяването има сходни характеристики с други неврологични заболявания. Само след задълбочен преглед на пациента лекарят може да направи окончателна диагноза.
Диагнозата предполага многостранно изследване на здравето на пациента, като се започне от събирането на анамнеза и завърши с молекулярно-генетичен анализ. Освен това в без провалневрологичен преглед, ЯМР, серологични и биохимични анализикръв. 
Какво трябва да бъде лечението?
В момента експертите не могат да предложат ефективни методилечение. Цялата помощ от лекарите се свежда до максимално улесняване на проявите на болестта.
Лечението на амиотрофична латерална склероза включва приемане на антихолинестеразни лекарства (Галантамин, Прозерин) за подобряване на качеството на речта и преглъщането, мускулни релаксанти (Диазепам), антидепресанти и транквиланти. При инфекциозни лезии се предписва антибиотична терапия. Кога силна болкалекарите препоръчват приема на нестероидни противовъзпалителни средства, впоследствие те се заменят с наркотични лекарства.
единственият ефективно лекарствоцеленасочено действие е Rilutek. Той не само удължава живота на пациента, но също така ви позволява да увеличите забавянето на прехвърлянето към вентилатора. 
Добрата грижа значително подобрява качеството на живот
Разбира се, всеки човек, който е диагностициран с ALS, се нуждае от правилна грижа. Пациентът разглежда критично състоянието си, защото всеки ден тялото му буквално избледнява. В крайна сметка такива хора престават да се обслужват самостоятелно, да общуват с роднини и приятели и изпадат в депресия.
Без изключение всички пациенти с ALS се нуждаят от:
- Във функционално легло със специален повдигащ механизъм.
- В тоалетната седалка.
- В инвалидна количка с автоматично управление.
- В средствата за комуникация, например в лаптоп.
Храненето на пациентите също изисква специално внимание. По-добре е да се дава добре поглъщана храна, богата на витамини и протеини. Впоследствие храненето без помощта на специална сонда не е възможно.
Амиотрофичната латерална склероза се развива бързо при някои хора. На роднини и приятели им е много трудно, защото човек буквално избледнява пред очите ни. Често хората, които се грижат за болни, се нуждаят от допълнителна помощ от психолог, както и от прием на успокоителни.
Прогноза
Ако лекарят потвърди амиотрофична латерална склероза, симптомите само се увеличават с всеки изминал ден, влошават се общо състояниепациент, прогнозата в този случай е разочароваща. В цялата история на съвременната медицина са регистрирани само два случая, когато пациентите са успели да оцелеят. За първия вече говорихме в тази статия. А вторият е известният физик Стивън Хокинг, който продължава успешно да съществува с такова заболяване през последните 50 години от живота си. Ученият работи активно и се радва на всеки нов ден, въпреки че се движи на специално оборудван стол и комуникира с другите чрез компютърен синтезатор на реч. 
Предпазни мерки
ОТНОСНО първична профилактикане е необходимо да се говори за патология, тъй като точните причини за появата му все още не са ясни. Вторичната профилактика трябва да е насочена към забавяне на прогресията на заболяването и удължаване на живота на пациента. Включва:
- Редовни консултации с невролог и прием на лекарства.
- Пълно отхвърляне на всички лоши навицитъй като те само обострят заболяването ALS.
- Лечението трябва да бъде адекватно и компетентно.
- Рационално и балансирано хранене.
Заключение
В тази статия говорихме за това какво представлява болестта ALS. Симптоми и лечение на това патологично състояниене трябва да се пренебрегва. За съжаление, съвременната медицина не може да предложи ефективна терапия срещу това заболяване. Роднините и приятелите обаче трябва да положат всички усилия за подобряване ежедневиеточовек с тази диагноза.
Амиотрофичната латерална склероза (ALS, „болест на Hehrig“, „болест на двигателния неврон“) е описана за първи път от френския психиатър Мартин Шарко през 1869 г.
В САЩ и Канада има още един термин - "болест на Лу Гериг", известен бейзболист, който трябваше да прекрати кариерата си на 36 години поради амиотрофична латерална склероза.
Какво е амиотрофична латерална склероза?
Амиотрофичната латерална склероза е заболяване на нервната система, което бързо засяга моторните неврони на гръбначния мозък, мозъчния ствол и кората.
IN патологичен процесучаствам двигателни нервичерепни неврони (лицеви, троични, глософарингеални).
Амиотрофичната латерална склероза е рядка (2-3 души на 100 000) и прогресира бързо.
В медицината има друга концепция - синдром на амиотрофична латерална склероза. Провокира се от друго заболяване, така че лечението в този случай е насочено към елиминиране на основната патология. Ако пациентът има симптоми на ALS, но причините за тях не са известни, лекарите не говорят за синдрома, а за болестта.
При ALS двигателните неврони се унищожават, те спират да изпращат сигнали от мозъка към мускулите, в резултат на което последните започват да отслабват и атрофират.
причини

Причините, които провокират появата на това заболяване, все още не са установени. Учените обаче предлагат няколко теории:
наследствена
Установено е, че в 10-15% от случаите заболяването е наследствено.
Вирусен
Тази теория получава широко разпространение през 60-те години на 20 век в САЩ и СССР. По това време бяха проведени експерименти върху маймуни. Животните са инжектирани с екстракти от гръбначния мозък на болни хора. Предполага се също, че заболяването може да бъде провокирано от полиомиелитния вирус.
Генная
Генно разрушаване се установява при 20% от пациентите с ALS. Те кодират ензима супероксиддисмутаза-1, който се оказва опасен за нервни клеткисупероксид към кислород.
автоимунни
Учените проведоха изследвания и откриха антитела, които убиват двигателните нервни клетки. Доказано е, че тези антитела могат да се образуват, когато тежки заболявания(Ходжкинов лимфом, рак на белия дроб и др.).
невронни
Тази теория е разработена от британски учени, които смятат, че образуването на ALS може да провокира елементи от глия - клетки, отговорни за жизнената активност на невроните. Ако функцията на астроцитите, които премахват глутамата от нервните окончания, е нарушена, рискът от болестта на Шарко се увеличава няколко пъти.
Сред рисковите фактори лекарите разграничават: наследствено предразположение, възраст над 50 години, тютюнопушене, работа, свързана с употребата на олово, военна служба.
Симптоми на ALS
Независимо от формата на заболяването, всички пациенти чувстват мускулна слабост, появяват се мускулни потрепвания и мускулната маса намалява.
Мускулната слабост нараства бързо, но очните мускули и сфинктера Пикочен мехурне са засегнати.
В ранен стадий на заболяването възниква:
- мускулна слабост в глезените и краката;
- атрофия на ръцете;
- нарушена моторика и реч;
- затруднено преглъщане;
- мускулни потрепвания;
- спазми на езика, ръцете и раменете.
С развитието на ALS се появяват пристъпи на смях и плач, нарушава се равновесието, появява се атрофия на езика.
Когнитивните функции се влошават само в 1-2% от случаите на заболяването, при други пациенти умствената дейност не се променя.
В по-късните етапи пациентът развива депресия, започват прекъсвания на дишането и се губи способността да се движи самостоятелно.
Болните от АЛС престават да се интересуват от близките си и от външния свят, стават капризни, необуздани, емоционално лабилни и агресивни. Когато дихателните мускули спрат да работят, човек се нуждае от механична вентилация.
Ход на заболяването
Първоначално се появяват симптоми, които пречат на човек да води пълноценен живот: мускулна изтръпване, конвулсии, потрепвания, затруднено говорене. Но, като правило, в самото начало е трудно да се определи точната причина за тези нарушения.
В повечето случаи ALS се поставя на етапа на мускулна атрофия.
Постепенно мускулната слабост се разпространява и обхваща нови части на тялото, пациентът не може да се движи самостоятелно, започват проблеми с дишането.
Пациентите с ALS рядко страдат от деменция, но тяхното състояние води до тежка депресия в очакване на смърт. В последния етап човек вече не може да се храни, ходи и диша самостоятелно, той се нуждае от специални медицински устройства.
Форми на заболяването
Формите на заболяването се отличават с местоположението на увредените мускули.
Булбарная
Засегнати са черепномозъчните нерви (9,10,12 двойки).
Пациентите с булбарна форма на ALS започват да имат проблеми с говора, оплакват се от затруднено произношение, трудно им е да движат езика си.
С прогресирането на заболяването актът на преглъщане се нарушава, храната може да се излее през носа. В късния стадий на заболяването мускулите на лицето и шията напълно атрофират, изражението на лицето изчезва и пациентите с ALS не могат да отварят устата си и да дъвчат храната.
цервико-торакален

Заболяването прогресира в горните крайници от двете страни.
Първоначално се появява дискомфорт в ръцете, за човек става трудно да извършва сложни движения с ръцете си, да пише, да свири на музикални инструменти. При преглед лекарят забелязва, че мускулите на ръцете на пациента са напрегнати и сухожилните рефлекси са повишени.
В напредналите стадии на заболяването мускулната слабост прогресира и се разпространява към предмишниците и раменете.
лумбосакрален
![]()
Първият симптом е слабост в долните крайници.
За пациента става по-трудно да върши работа, докато стои, да се изкачва по стълби, да кара велосипед и да ходи на дълги разстояния.
С течение на времето стъпалото започва да увисва, походката се променя, след това мускулите на краката напълно атрофират, човекът не може да ходи, развива се инконтиненция на урина и изпражнения.
Почти 50% от пациентите страдат от цервико-торакална форма на ALS, по 25% са лумбосакрални и булбарни.
Диагностика
Неврологът използва като основни диагностични методи:
MRI на главния и гръбначния мозък
С помощта на този метод е възможно да се открие дегенерация на пирамидални структури и атрофия на двигателните части на мозъка.
Неврофизиологични изследвания
За откриване на ALS се използват TKMS (транскраниална магнитна стимулация), ENG (електроневрография), EMG (електромиография).
Цереброспинална пункция
Определя се нивото на протеиново съдържание (нормално или повишено).
Биохимични изследвания на кръвта
При пациенти с ALS се установява повишаване на креатинфосфокиназата 5 или повече пъти, натрупване на креатинин и урея и повишаване на AST и ALT.
Молекулярно-генетичен анализ
Генът, кодиращ супероксид дисмутаза-1, се изследва.
Но тези методи не са достатъчни за идентифициране на диагнозата, паралелно използвани диференциална диагноза за потвърждаване или изключване на заболявания:
- мозък: дисциркулаторна енцефалопатия, тумори на задната черепна ямка, мултисистемна атрофия.
- гръбначен мозък: тумори, сирингомиелия, лимфоцитна левкемия, спинална амиотрофия и др.
- мускули: миозит, окулофарингеална миодистрофия, миотония Rossolimo-Steiner-Kurshman.
- периферни нерви: мултифокална моторна невропатия, синдром на Parsonage-Turner и др.
- нервно-мускулен синапс: Синдром на Lambert-Eaton, миастения гравис.
Лечение
Няма лечение за ALS, но е възможно да се забави прогресията на това заболяване, да се увеличи продължителността на живота и да се облекчи състоянието на човек.
За това се използва комплексна терапия:
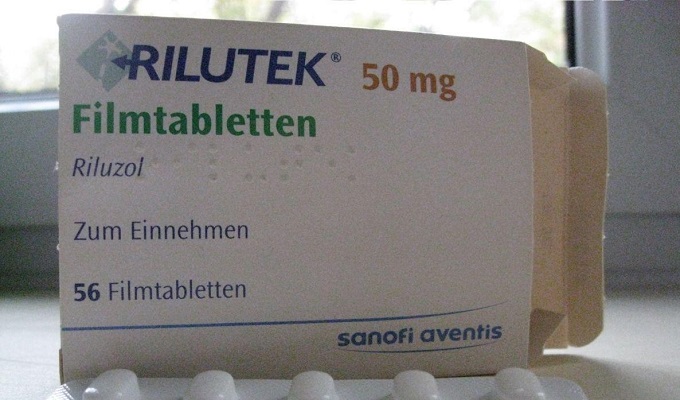
Лекарството, което за първи път е използвано при лечението на ALS в Обединеното кралство и САЩ. Активните вещества блокират освобождаването на глутамин и забавят процеса на увреждане на невроните. Лекарството трябва да се приема 2 пъти на ден за 0,05 g.
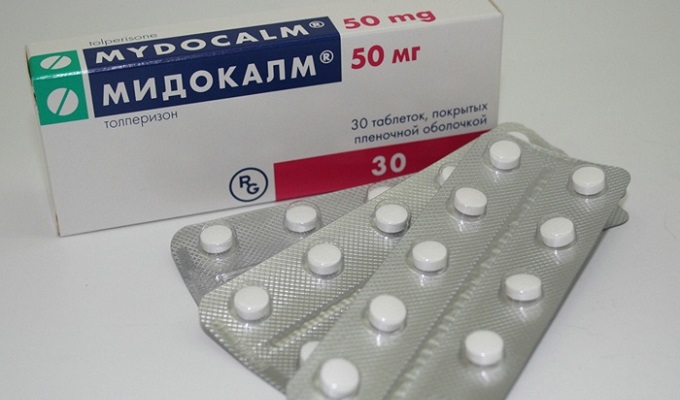
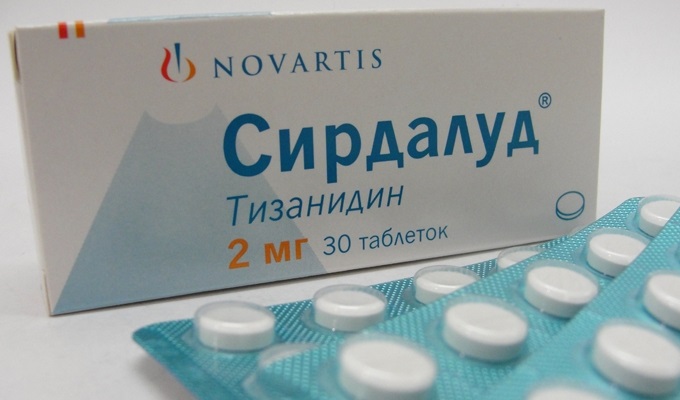
Мускулни релаксанти и антибиотиципомощ при мускулна слабост. За премахване на мускулни спазми и потрепвания се предписват Mydocalm, Baclofen, Sirdalud.

За увеличаване мускулна масаизползва се анаболен "Retabolin".
Антибиотици, ако се развие сепсис или инфекциозни усложнения. Лекарите предписват флуорохинолони, цефалоспорини, карбопени.
витаминигрупи B, E, A, C за подобряване на импулса по нервните влакна.
Антихолинестеразни лекарства, които забавят процеса на разрушаване на ацетилхолина ("Калимин", "Прозерин", "Пиридостигмин").

В някои случаи се използва трансплантация на стволови клетки. Предотвратява смъртта на нервните клетки, насърчава растежа на нервните влакна и възстановява невронните връзки.
В по-късните етапи използвайте антидепресанти и транквиланти, нестероидни болкоуспокояващи и опиати.
Ако сънят е нарушен, се предписват бензодиазепинови препарати.
За улесняване на движението столовете и леглата с различни функции, бастуни, фиксиращи яки. Лекарите съветват логопедична терапия. В по-късните стадии на заболяването ще е необходим ежектор за слюнка, а след това и трахеостомия, за да може пациентът да диша.
Нетрадиционните методи за лечение на ALS не дават положителен резултат.
Прогноза и последствия
Прогноза за пациенти с ALS неблагоприятен. Летален изход настъпва за 2-12 години, като тежка пневмония, дихателна недостатъчност или др. сериозно заболяванепричинени от болестта на Gehrig.
При булбарна форма и при пациенти в напреднала възраст периодът се намалява до 3 години.
Предотвратяване
Мерките за предотвратяване на ALS лекарството все още не са известни.
Заболяването прогресира много бързо, няма случаи на успешно лечение и възстановяване на двигателните функции на тялото. Мускулната слабост, която се увеличава постепенно, напълно променя живота на човек и семейството му.
Но въпреки разочароващата прогноза и недостатъчното проучване на болестта, близките трябва да се надяват, че в близко бъдеще ще бъдат разработени ефективни лечения. терапевтични методилечение. Междувременно е необходимо да се вземат мерки за облекчаване на състоянието на човек с ALS.
амиотрофична латерална склероза - опасна болесткоето може да обездвижи човек и да доведе до смърт.
Лекарите все още не могат да установят точните причини за заболяването и да намерят ефективно лечение. В момента единственото, което медицината може да направи, е да облекчи състоянието на пациентите с ALS. Нито един пациент не може да се излекува напълно от това заболяване. Важно е да се разграничи "ALS заболяване" от "ALS синдром". Във втория случай прогнозата за възстановяване е много по-добра.
(ALS), известна още като болестта на Лу Гериг, е бавно прогресиращо, нелечимо дегенеративно заболяване на централната нервна система. Според Щатската асоциация само половината от жителите на САЩ са чували за болестта, същият модел се наблюдава и в други страни.
Един от начините да привлечете внимание на проблема с амиотрофичната латерална склерозасе превърна в Ice Bucket Challenge, в който хората трябва да се полеят с кофа с ледена вода и да направят дарение. През август 2014 г. кампанията придоби особена популярност по света, като успя да привлече 50 милиона долара дарения и повече от 1,5 милиона участници. президент и изпълнителен директоркомпания 3 M Inge Thulin се присъедини към участниците и коментира участието си в акцията:
"амиотрофична латерална склероза(БАН) — ужасна болест. Приех предизвикателство от семейството на нашия служител на 3M Алън Уолгрен, който страда от това заболяване повече от 32 години. Той беше диагностициран в началото на годината, а днес вече е почти напълно парализиран. Точно преди една година загубихме и един от най-добрите лидери в денталния бизнес на 3M, Лари Лиър, който почина от ALS. Видях колко бързо "изгоря", беше ужасно. И приех това предизвикателство не само в чест на Алън и Лари, но и в чест на всички семейства, които са изправени пред тази ужасна болест."
Причини за заболяването ALS
Причината за ALS е мутация на някои протеини (убиквитин) с появата на вътреклетъчни агрегати. Семейните форми на заболяването се наблюдават в 5% от случаите. По принцип заболяването ALS засяга хора на възраст над четиридесет и шестдесет години, от които не повече от 10% са носители на наследствената форма, учените все още не могат да обяснят останалите случаи с влиянието на който и да е външни влияния- екология, наранявания, заболявания и други фактори.
Симптоми на заболяването
Ранните симптоми на заболяването са конвулсии, потрепвания, изтръпване и слабост в крайниците, както и затруднения в говора, но подобни признаци се отнасят за голям брой заболявания. Това го прави много трудно да се диагностицира до последния период, болестта вече преминава в етапа на мускулна атрофия.
Първоначални ALS лезии могат да възникнат на различни частитялото, докато до 75% от пациентите заболяването започва с крайниците, главно долните.
Какво е? Как се проявява
Първоначални прояви на заболяването:
.слабост в дисталните части на ръцете, неловкост при извършване на фини движения с пръстите, загуба на тегло в ръцете и фасцикулации (мускулни потрепвания)
.по-рядко заболяването дебютира със слабост в проксималните ръце и раменния пояс, атрофия на мускулите на краката в комбинация с долна спастична парапареза
Възможно е и началото на заболяването с булбарни нарушения - дизартрия и дисфагия (25% от случаите)
Крампите (болезнени контракции, мускулни спазми), често генерализирани, се появяват при почти всички пациенти с ALS и често са първият признак на заболяването
Характеристика клинични проявленияБАС
Амиотрофичната латерална склероза се характеризира с комбинирано увреждане на долния двигателен неврон (периферен) и увреждане на горния двигателен неврон (питамидни пътища и / или пирамидални клетки на моторния кортекс на мозъка).
Признаци на увреждане на долния двигателен неврон:
- мускулна слабост (пареза)
- хипорефлексия (намален рефлекс)
- мускулна атрофия
- фасцикулации (спонтанни, бързи, неритмични контракции на снопове мускулни влакна)
Признаци на увреждане на горния двигателен неврон:
- мускулна слабост (пареза).
- спастичност (повишен мускулен тонус)
- хиперрефлексия (повишени рефлекси)
- патологични признаци на краката и ръцете
За ALS в повечето случаи асиметрия на симптомите.
В атрофирали или дори външно непокътнати мускули, фасцикулации(мускулни потрепвания), които могат да бъдат в локална мускулна група или да са широко разпространени.
IN типичен случайначалото на заболяването със загуба на тегло на тенарните мускулиедна от ръцете с развитие на слабост на аддукцията (привеждане) и противопоставяне на палеца (обикновено асиметрично), което затруднява хващането с палеца и показалцитеи води до нарушения на контрола на фината моторика в мускулите на ръката. Болният изпитва затруднения при взимане на малки предмети, при закопчаване на копчета, при писане.
След това, когато болестта прогресира, мускулите на предмишницата участват в процеса и ръката придобива вид на „лапа с нокти“. Няколко месеца по-късно се развива подобна лезия на другата ръка. Атрофия, която постепенно се разпространява, улавя мускулите на рамото и раменния пояс.
По същото време или по-късночесто се развива увреждане на булбарните мускули: фасцикулации и атрофия на езика, пареза на мекото небце, атрофия на мускулите на ларинкса и фаринкса, което се проявява под формата на дизартрия (нарушения на говора), дисфагия (нарушения на преглъщането), отделяне на слюнка.
Мимически и дъвкателни мускулиобикновено се засягат по-късно от другите мускулни групи. С развитието на болестта става невъзможно изпъкването на езика, издуването на бузите и опъването на устните в тръба.
Понякога се развива слабост на екстензорите на главатапоради което пациентът не може да държи главата си изправена.
При участие в процеса на диафрагматанаблюдава се парадоксално дишане (при вдъхновение стомахът потъва, при издишване изпъква).
Обикновено първо атрофират кракатапредни и странични мускулни групи, което се проявява с „висящо стъпало“ и походка тип стъпка (пациентът повдига високо крака си и го хвърля напред, рязко го спуска).
Характерно е, че мускулната атрофия е селективна.
- На ръцете се наблюдава атрофия:
тенара
хипотенар
междукостни мускули
делтоидни мускули
- На краката участват мускулите, които извършват дорзалната флексия на стъпалото.
- В булбарните мускули се засягат мускулите на езика и мекото небце.
пирамидален синдромразвива се, като правило, в ранен стадий на ALS и се проявява чрез съживяване на сухожилните рефлекси. След това често се развива долна спастична парапареза. В ръцете повишаването на рефлексите се комбинира с мускулна атрофия, т.е. има комбинирана, едновременна лезия на централните (пирамидални) пътища и периферния двигателен неврон, което е характерно за ALS. Повърхностните коремни рефлекси изчезват с напредване на процеса. Симптомът на Бабински (с пунктирано дразнене на стъпалото, големият пръст се разгъва, другите пръсти се разминават и разгъват във формата на ветрило) се наблюдава в половината от случаите на заболяването.
Възможно е да има сензорни нарушения. При 10% от пациентите се наблюдават парестезии в дисталните части на ръцете и краката. Болката, понякога силна, обикновено нощна, може да бъде свързана със скованост на ставите, продължителна неподвижност, спазми поради висока спастичност, с крампи (болезнени мускулни спазми), депресия. Загубата на чувствителност не е типична.
Окуломоторни нарушенияне са характерни и се появяват в крайните стадии на заболяването.
Дисфункцията на тазовите органи не е типична, но в напреднал стадий може да се появи задържане на урина или инконтиненция.
Умерено когнитивно увреждане(намаляване на паметта и умствената работоспособност) се проявяват при половината от пациентите. При 5% от пациентите се развива фронтален тип, който може да се комбинира с паркинсонизъм.
Характеристика на ALS е липсата на рани от залежаване дори при парализирани лежащо болни.
Където и да се появят първи признаци на ALS, мускулната слабост постепенно се прехвърля в по-големи части на тялото, въпреки че при булбарната форма на ALS пациентите може да не оцелеят до пълна пареза на крайниците поради спиране на дишането.
С течение на времето пациентът губи способността си да се движи самостоятелно. ALS заболяванене засяга умственото развитие, но най-често започва дълбока депресияЧовекът чака смъртта. В крайните стадии на заболяването се засягат и мускулите, които изпълняват дихателната функция, животът на пациентите трябва да се поддържа чрез изкуствена вентилация на белите дробове и изкуствено хранене. От забелязването на първите признаци на ALS до смъртта са необходими 3-5 години. Въпреки това, случаите са широко известни, когато състоянието на пациенти с недвусмислено разпознато заболяване ALS се стабилизира с течение на времето.
КОЙ ИМА АЛС?
В света има повече от 350 000 пациенти с ALS.
ALS се диагностицира при 5-7 души годишно на 100 000 души население. повече от 5600 американци са диагностицирани с ALS всяка година. Това са 15 нови случая на Bass на ден
ALS може да засегне всеки. Процент на заболеваемост (брой нови) ALS - 100 000 души годишно
По-малко от 10% от случаите на ALS са наследствени. ALS може да засегне както мъжете, така и жените ALS засяга всички етнически и социално-икономически групи
ALS може да засегне млади или много възрастни възрастни, но най-често се диагностицира в средна и късна възраст.
Хората с ALS се нуждаят от скъпо оборудване, лечение и постоянна грижа 24/7
90% от тежестта на грижите пада върху плещите на членовете на семейството на пациентите с ALS. ALS води до евентуално отслабване на физическото, емоционалното и финансови ресурсиИма повече от 8500 пациенти с ALS в Русия и повече от 600 пациенти с ALS в Москва, въпреки че този брой официално постоянно се подценява. Най-известните руснаци с ALS са Дмитрий Шостакович, Владимир Мигуля.
Причините за заболяването са неизвестни. Няма лек за ALS. Имаше забавяне на хода на заболяването. Удължаването на живота е възможно с помощта на домашен вентилатор.
Синдромите, които са клинично неразличими от класическия ALS, могат да бъдат резултат от:
Структурни лезии:
парасагитални тумори
форамен магнум тумори
спондилоза цервикаленгръбначен стълб
Синдром на Арнолд-Киари
хидромиелия
артериовенозна аномалия на гръбначния мозък
Инфекции:
бактериални - тетанус, лаймска болест
вирусни - полиомиелит, херпес зостер
ретровирусна миелопатия
Интоксикации, физически агенти:
токсини - олово, алуминий, други метали.
лекарства - стрихнин, фенитоин
токов удар
рентгенови лъчи
Имунологични механизми:
плазмоцитна дискразия
автоимунна полирадикулоневропатия
Паранеопластични процеси:
паракарциноматозни
паралимфоматозен
Метаболитни нарушения:
хипогликемия
хиперпаратироидизъм
тиреотоксикоза
дефицит на фолат,
витамини В12, Е
малабсорбция
Наследствени биохимични нарушения:
дефект на андрогенния рецептор - болест на Кенеди
дефицит на хексозаминидаза
дефицит на а-глюкозидаза - болест на Помпе
хиперлипидемия
хиперглицинурия
метилкротонилглицинурия
Всички тези състояния могат да причинят симптомите, наблюдавани при ALS, и трябва да се имат предвид при диференциалната диагноза.
Няма ефективно лечение на заболяването. Единственото лекарство, инхибиторът на освобождаване на глутамат рилузол (Rilutek), забавя смъртта с 2 до 4 месеца. Предписва се по 50 mg два пъти дневно.
Лечение на заболяването ALS
Основата на лечението е симптоматична терапия:
- Физиотерапия.
Физическа дейност. Пациентът трябва, доколкото е възможно, да поддържа физическа дейностС напредването на болестта има нужда от инвалидна количка и други специални устройства.
.Диета. Дисфагията създава опасност от навлизане на храна в дихателните пътища. Понякога има нужда от храна през сонда или в гастростома.
- Използването на ортопедични средства: цервикална яка, различни шини, устройства за захващане на предмети.
С крампи (болезнени мускулни спазми): хинин сулфат 200 mg два пъти дневно, или фенитоин (дифенин) 200-300 mg / ден, или карбамазепин (Finlepsin, Tegretol,) 200-400 mg / ден, и / или витамин Е 400 mg два пъти дневно, както и като магнезиеви препарати, верапамил (Isoptin).
.При спастичност: баклофен (Baclosan) 10 - 80 mg / ден или тизанидин (Sirdalud) 6 - 24 mg / ден, както и клоназепам 1 - 4 mg / ден или мемантин 10 - 60 mg / ден.
- За слюноотделяне атропин 0,25 - 0,75 mg три пъти дневно или хиосцин (Buscopan) 10 mg три пъти дневно.
Ако е невъзможно да се яде поради нарушение на преглъщането, се прилага гастростомия или се поставя назогастрална сонда. Ранната перкутанна ендоскопска гастростома удължава живота на пациентите средно с 6 месеца.
- При болкови синдромиизползвайте целия арсенал от аналгетици. Включително наркотични аналгетици в крайните стадии.
- Понякога известно временно подобрение се постига от антихолинестеразни лекарства (неостигмин метилсулфат - прозерин).
Cerebrolysin във високи дози (10-30 ml IV капково за 10 дни в повтарящи се курсове). Има редица малки проучвания, показващи невропротективната ефикасност на церебролизин при ALS.
.Антидепресанти: Sertalin 50 mg/ден или Paxil 20 mg/ден или Amitriptyline 75-150 mg/ден странични ефекти- причинява сухота в устата, следователно намалява хиперсаливацията (саливация), която често измъчва пациенти с АЛС).
.При респираторни нарушения: изкуствена вентилациябелите дробове в болнични условия, като правило, не се извършва, но някои пациенти купуват преносими вентилатори и извършват механична вентилация у дома.
- В ход са разработки за използването на растежен хормон, невротрофични фактори при ALS.
- Напоследък активно се развива терапията със стволови клетки. Този метод обещава да бъде обещаващ, но все още е на етап научни експерименти.
лекарства, лекарства, които помагат за забавяне на заболяването ALS:
Съвременната медицина непрекъснато се развива. Учените създават нови лекарства за нелечими досега заболявания. Днес обаче специалистите не могат да предложат адекватно лечение срещу всички заболявания. Една от тези патологии е болестта ALS. Причините за това заболяване все още не са проучени и броят на пациентите се увеличава всяка година. В тази статия ще разгледаме по-подробно тази патология, нейните основни симптоми и методи на лечение.
Главна информация
Амиотрофичната латерална склероза (ALS, болест на Шарко) е сериозна патология на нервната система, при която има увреждане на така наречените двигателни неврони в гръбначния мозък, както и в кората на главния мозък. Това е хронично и нелечимо заболяване, което постепенно води до дегенерация на цялата нервна система. В последните стадии на заболяването човек става безпомощен, но в същото време запазва умствена яснота и психическо здраве.
Болестта на ALS, чиито причини и патогенеза не са напълно проучени, не се различава по специфични методи за диагностика и лечение. Учените продължават активно да го изучават и днес. Сега е известно със сигурност, че заболяването се развива главно при хора на възраст от 50 до около 70 години, но има случаи на по-ранни лезии.
Класификация
В зависимост от локализацията на първичните прояви на заболяването, експертите разграничават следните форми:
- Лумбосакрална форма (има нарушение на двигателната функция на долните крайници).
- Булбарна форма (някои ядра на мозъка са засегнати, което води до характер).
- Цервико-торакална форма ( първични симптомисе проявява с промени в обичайната двигателна функция Горни крайници).
От друга страна, експертите разграничават още три вида заболяване на ALS:
Защо възниква ALS?
причини тази болестза съжаление все още остават слабо разбрани. В момента учените идентифицират редица фактори, при наличието на които вероятността от заболяване се увеличава няколко пъти:
ALS заболяване. Симптоми
Снимки на пациенти с това заболяване могат да се видят в специализирани справочници. Всички те имат само едно общо нещо - външни симптомизаболяване в по-късните етапи.
Що се отнася до основното клинични признаципатологии, те много рядко предизвикват бдителност от страна на пациентите. Освен това потенциалните пациенти често ги обясняват с постоянен стрес или липса на почивка от работната рутина. Следните симптоми на заболяването се появяват в ранните етапи:
- мускулна слабост;
- дизартрия (затруднено говорене);
- чести мускулни спазми;
- изтръпване и слабост на крайниците;
- леки мускулни потрепвания.
Всички тези признаци трябва да предупредят всички и да станат причина да се свържете с специалист. В противен случай заболяването ще прогресира, което няколко пъти увеличава вероятността от усложнения.
Протичането на заболяването
Как се развива ALS? Заболяването, чиито симптоми са изброени по-горе, първоначално започва с мускулна слабости изтръпване на крайниците. Ако патологията се развие от краката, тогава пациентите могат да изпитват затруднения при ходене, постоянно се спъват.
Ако заболяването се прояви с нарушение на горните крайници, има проблеми с изпълнението на най-елементарните задачи (закопчаване на риза, завъртане на ключа в ключалката). 
Как иначе може да се разпознае ALS? Причините за заболяването в 25% от случаите се крият в лезията продълговатия мозък. Първоначално има затруднения с говора, а след това и с преглъщането. Всичко това води до проблеми с дъвченето на храната. В резултат на това човек спира да се храни нормално и отслабва. В резултат на това много пациенти попадат в депресия, тъй като заболяването обикновено не засяга
Някои пациенти имат затруднения с формирането на думи и дори с нормалната концентрация. Незначителните нарушения от този вид най-често се обясняват с лошо дишане през нощта. Медицински работницивече трябва да разкажат на пациента за характеристиките на хода на заболяването, възможностите за лечение, за да може предварително да вземе информирано решение за бъдещето си.
Повечето пациенти умират от дихателна недостатъчностили пневмония. По правило смъртта настъпва в рамките на пет години от датата на потвърждаване на заболяването.
Диагностика
Само специалист може да потвърди наличието на това заболяване. В този случай основната роля се дава на компетентното тълкуване на съществуващото клинична картинапри конкретен пациент. Диференциалната диагноза на заболяването ALS е изключително важна.
Какво трябва да бъде лечението?
За съжаление медицината днес не може да предложи ефективна терапия срещу това заболяване. Как може да се излекува ALS? Лечението трябва да бъде насочено предимно към забавяне на хода на патологията. За тези цели се използват следните мерки:

Лечение със стволови клетки
В много европейски страни пациентите с ALS вече се лекуват със собствени стволови клетки, което също забавя прогресията на заболяването. Този вид терапия е насочена към подобряване на основните функции на мозъка. трансплантирани в зоната на увреждане, възстановяват невроните, подобряват снабдяването на мозъка с кислород и насърчават появата на нови кръвоносни съдове.
Изолирането на самите стволови клетки и тяхната директна трансплантация, като правило, се извършват амбулаторно. След терапията пациентът остава в болницата още 2 дни, където специалисти проследяват състоянието му. 
По време на самата процедура се инжектират клетки в гръбначно-мозъчна течностте нямат право да се възпроизвеждат сами чрез Извън тялото, а реимплантирането се извършва само след детайлно почистване.
Важно е да се отбележи, че този вид терапия може значително да забави заболяването ALS. Снимки на пациенти 5-6 месеца след процедурата ясно доказват това твърдение. Това няма да помогне да се отървете напълно от болестта. За съжаление има и случаи, когато лечението не дава никакъв ефект.
Заключение
Прогнозата на това заболяване почти винаги е неблагоприятна. Прогресия двигателни нарушениянеизбежно води до (2-6 години).
В тази статия говорихме за това какво представлява такава патология като ALS. Заболяване, чиито симптоми може да не се появят дълго време, в момента е невъзможно да се излекува напълно. Учените от цял свят продължават активно да изучават това заболяване, причините и скоростта на развитие, опитвайки се да намерят ефективно лекарство.

