НАСЛЕДСТВЕНИ БОЛЕСТИ НА НЕРВНАТА СИСТЕМА
ЛЕКЦИЯ 16
Дегенеративните заболявания, засягащи предимно нервно-мускулната система, представляват най-голямата група от всички наследствени заболявания.
Изключително важни и често решаващи в диагностиката на нервно-мускулните заболявания са резултатите от електрофизиологичните и биохимични изследвания. Също толкова голямо е значението на патоморфологичните находки. Изследването на материал от мускулна биопсия под светлинен микроскоп помага да се разграничи миогенната атрофия от неврогенната атрофия. Хистохимичното изследване е необходимо за идентифициране на метаболитни мускулни лезии и електронна микроскопияоткри цял голям клас заболявания - непрогресивни миопатии.
Лекото разтягане на засегнатите стави намалява появата на контрактури и по този начин води до по-бавно прогресиране на заболяването. В този случай се изпълняват различни специфични упражнения, а притискащата превръзка Urias се оказа ефективна в случаи на нарушена дълбочина на крайниците и често се счита за домашно лечение за изписване. Сензомоторното обучение за пациенти с мускулни нарушения се провежда в индивидуална или групова терапия и се фокусира върху координацията и фината моторика.
Много важен методЛечението при хора с мускулни заболявания е възстановяване на способността за шофиране. За тази цел има сътрудничество с външна школа за шофиране в отделението по трудова терапия, а шофирането в Бад Вилдунген се извършва в подходящо превозно средство за хора с увреждания с обучен инструктор по шофиране.
Прогресивна мускулна дистрофия.Терминът мускулни дистрофии се отнася до група от генетично обусловени заболявания, характеризиращи се с прогресиращо развитие дегенеративни променив мускулни влакна без първична патология на периферния (долния) двигателен неврон.
Различните форми се различават една от друга по видовете наследство, времето на началото на процеса, характера и скоростта на протичане, уникалната топография на мускулната атрофия, наличието или отсъствието на псевдохипертрофия и ретракции на сухожилията и други характеристики .
Допълнителен подход към рехабилитацията на мускулни нарушения е осигуряването на адекватно консултиране. Това се отнася не само за ортезите, но и за всички останали помощни средствау дома, Целевата помощ за хора с увреждания с мускулни смущения е изключително полезна, инвалидните колички са прецизно измерени и приспособени към нуждите на хората с мускулни смущения. Душ стол, асансьор за баня, инвалидни колички, рампи за инвалидни количкии други.
Отделението по физикална терапия използва различни процедури за релаксация на пациента. В допълнение, този клон на лечение може да намали често незначителните симптоми на болка при пациенти с мускулни нарушения. Логопедията е друг клон на рехабилитацията за лечение на мускулни нарушения. Тук също трябва да се постави друга диагноза с ларингоскопия или определено Рентгенови изследвания. В квалифицирана рехабилитационна клиника за лечение на мускулни заболявания, холистичният терапевтичен подход към това заболяване е важна предпоставка.
Повечето мускулни дистрофии са добре проучени клинично и Подробно описаниенаправени в края на миналия век. Но въпреки почти вековната история на изучаване на мускулните дистрофии, въпросите за тяхната патогенеза и лечение остават нерешени и до днес. Големи надежди се възлагат на молекулярната генетика, с помощта на която е определено местоположението на гените за много нозологични форми.
Това включва завършили студенти по психология от катедрата. Засегнатите лица страдат от физически дефицити и свързаните с тях последици по-специално - Продължителността на живота често се свързва с депресивни настроенияи психосоматични оплаквания. Поддържащите индивидуални разговори или изучаването на техники за релаксация като автогенно обучение или прогресивна мускулна релаксация са много полезни тук.
Въпроси относно закона за кърмене, строго увреждане, работно място, контакт с работодателя за професионална рехабилитация или интеграция в компанията, трябва да бъдат споменати тук като чести теми в социалното консултиране. В допълнение, целенасоченото консултиране в консултациите е често често срещан въпросза пациенти с мускулни заболявания. И накрая, планът за лечение по време на рехабилитация в Бад Вилдунген включва и координация от квалифициран специалист ортопед. Wiezerek е главен лекар на ортопедичния отдел в клиниката Homberg в Бад Вилдунген.
Диагнозата на мускулна дистрофия често е много трудна. Има голяма вариабилност в клиничните прояви, а малкият брой членове на семейството затруднява определянето на начина на унаследяване.
Характерен двигателен дефект при пациенти с мускулна дистрофия е "патешка" походка: пациентът ходи, клатушкайки се от една страна на друга. Свързва се главно със слабост на седалищните мускули, предимно средните и малките мускули, които фиксират таза спрямо бедрената кост. В резултат на това заболяването причинява накланяне на таза към неопорния крак (феномен на Тренделенбург) и компенсаторно накланяне на торса към противоположната страна(феноменът на Дюшен). При ходене страната на наклона постоянно се променя. Тези промени могат да бъдат проверени и чрез теста на Тренделенбург, като помолите пациента да повдигне единия си крак, сгъвайки го под прав ъгъл в коленните и тазобедрените стави: тазът от страната на повдигнатия крак се спуска (и не се повдига както обикновено) поради слабост на глутеус медиус мускул на опорния крак.
Клиниката провежда регулярни обучения, както и участие в съответни конгреси и симпозиуми. Тази среща се състоя в частност. Често се наблюдава и повишаване на мускулните ензими. Болестите обикновено се развиват бавно, но възникват бързи заболявания. Ако подозирате, че имате миопатия или имате известно невромускулно заболяване, можете да насрочите срещи.
Лечението зависи от конкретна причина, възпалително мускулно заболяване може, например, Например, различни имунотерапии. Детайлен прегледРазнообразие от генетични мускулни заболявания могат да бъдат намерени в таблиците на гените на невромускулните заболявания.
Издигайки се от хоризонтално положение, пациент с тежка мускулна слабост на проксималните мускули почти не се обръща по корем, след това, опирайки ръцете си на пода, става на четири крака и след това, опирайки ръцете си на пищялите, след това на бедрата , постепенно се изправя. Този феномен на „печелене сам“ се нарича маневра на Говърс. Често се свързва със слабост на глутеус максимус мускулите.
Лекарства, които могат да причинят или засилят мускулно заболяване
Всички необходими диагностични възможности са налични в нашата практика. Това включва практика и редовно обучение в областта на нервно-мускулните заболявания. Засегнатите могат да се регистрират и в пациентския регистър. Регистрите на пациентите са важна първа стъпка към по-доброто разбиране на болестите и изследванията в областта на терапевтичното развитие.
Увреждане на сърдечния мускул
Тук се разглежда възможното участие на сърдечния мускул в миопатии и други невромускулни заболявания.Мускулна дистрофия на Дюшен.Псевдохипертрофичната мускулна дистрофия на Дюшен се среща по-често от всички други заболявания на мускулната система (30 на 100 000 живи новородени). Характеризира се с ранно начало и злокачествено протичане. Класическата картина се проявява чрез промяна в походката на дете на възраст 2-5 години; на възраст 8-10 години децата вече ходят трудно; на 14-15 години те са като правило, напълно обездвижен. Децата имат повече ранна възрастпървоначалните симптоми се проявяват чрез забавено двигателно развитие: те започват да ходят по-късно и не могат да тичат или скачат. Пациентите умират през 2-3-то десетилетие от живота.
Имаме предвид всички нервно-мускулни заболявания. Според класификацията на Уолтън има 800 форми. Този симптом може да бъде ограничен до няколко мускулни групи или, в зависимост от заболяването, може да присъства цялата мускулатура. Мускулна загуба, намаление мускулна масаИ мускулна слабостса основните симптоми на невромускулни заболявания. Лекарствата се наричат намаляване на мускулната атрофия. Те обаче все още не дават директна диагноза. Има редица причини, които могат да доведат до частична загуба на мускулна маса, която отчасти е свързана с цялото тяло.
Един от първите признаци на заболяването е удебеляване на мускулите на прасеца и постепенно увеличаване на обема им поради псевдохипертрофия. Атрофията на мускулите на бедрото и тазовия пояс често се маскира от добре развита подкожна мастна тъкан. Постепенно процесът поема възходяща посока и се разпространява извън раменния пояс, мускулите на гърба и след това към проксималните части на ръцете.
Истинската причина за такава мускулна атрофия или мускулна слабост може да се крие в отдалечени, напълно различни области на тялото. Причините могат да се крият в нервните клетки гръбначен мозък, отговорен за движението, в захранващите нерви, при прехвърляне на нерв към мускул или в самите мускули.
Лечение на нервно-мускулни заболявания
Има обещаващи лекарства за лечение на миозит, миастения гравис и ендокринна миопатия. Първите подходи за забавяне са възможни с латерална склерозаамиотрофия. В случай на наследствени мускулни дистрофии и спинални мускулни атрофии, причинно-следственото лечение все още не е установено. Насочени са към конкретни очаквания за бъдещето генна терапия. При всички невромускулни заболявания симптоматиката се състои от последователна физиотерапевтична подкрепа в комбинация с ортопедични мерки, в някои случаи с дихателна подкрепа и в някои случаи с възможност за сърдечна трансплантация.
IN терминален стадиймускулната слабост може да се разпространи към мускулите на лицето, фаринкса и дихателните мускули.
В напреднал стадий на заболяването има такива характерни симптоми, как " патешка разходка”; подчертана лумбална лордоза, крилати лопатки, симптом на "отпуснат раменен пояс". Характерни са ранните мускулни контрактури и ретракции на сухожилията, особено на ахилесовото сухожилие. Коленните рефлекси изчезват рано, а след това и рефлексите от горните крайници.
Лечението включва и компенсиране на физическите ограничения с адаптации. Германското дружество за мускулни заболявания създаде независим ресурсен център. Има и възможност да тествате продуктите у дома в два стандартни апартамента или да прекарате само няколко дни в апартаменти без бариери.
Мускулни нарушения Независими нарушения Мускулни сравнително редки. От друга страна, често се случва мускулите да бъдат засегнати от други основни заболявания, особено болести нервна система; Инфекциозни заболяваниякато тиф и туберкулоза; Паразитни заболявания, тринити и свински глисти, както и хормонални нарушения.
Псевдохипертрофията може да се развие не само в мускулите на прасеца, но и в глутеалните, делтоидните, коремните и езиковите мускули. Много често сърдечният мускул страда от вид кардиомиопатия. Нарушения в ритъма на сърдечната дейност, разширяване на границите на сърцето, глухота на тоновете, ЕКГ промени. Острата сърдечна недостатъчност е най обща причиналетален изход при мускулна дистрофия на Дюшен. При аутопсия се установява фиброза и мастна инфилтрация на сърдечния мускул.
Обикновената мускулна загуба често се случва в резултат на консервация или почивка Мускулната контракция може да бъде причинена от сраствания или наранявания поради лоша циркулация - В резултат на удушаване на асоциациите. Причината за мускулна болка може да бъде мускулен ревматизъм, пренапрежение, простуда, метаболитни нарушения или пренапрежение на определени мускулни групи поради деформация на скелета.
миома - доброкачествен тумор мускулна тъкан. Полимиозит: мускулно заболяване със симптоми, подобни на дерматомиозит, но без кожен обрив. Що се отнася до причината, предполага се, че разстройството се отнася до автоагресиите, като страдание, което идва в резултат на това, че тялото атакува собствените си структури.
Често се наблюдават нарушения на стомашно-чревния мотилитет.
Често срещан симптом е намалената интелигентност. Интересно е, че в някои семейства умствената изостаналост е изразена, в други е сравнително умерена. Промените във висшите психични функции обикновено не прогресират и не корелират с тежестта на мускулния дефект. Не може да се обясни само с педагогическото занемаряване на болните деца, които рано биват изключвани от детски групи и не ги посещават. детска градинаи училище поради двигателни дефекти. CT и MRI често разкриват церебрална атрофия, вероятно свързана с нарушено пренатално развитие на мозъка.
Има повече от 200 форми на мускулни заболявания, някои наречени на техните откриватели, а други на разстройството. Най-често срещаните са три подгрупи. Прогресивна мускулна дистрофия Спинална мускулна атрофия Неврална мускулна атрофия. В клиничната практика тези заболявания са редки и са абсолютно необходими в ръцете на невролог.
Генетичният дефект на хромозома 19 е увреждане мускулни клетки, които не могат да бъдат лекувани и до днес. Причината вероятно е промяна в мембранната система на мускулните клетки. Мистичната дистрофия може да прогресира до пълно унищожаване на мускулните клетки. Първо се засягат мускулите на лицето, ръцете, предмишниците, долната част на краката и стъпалата. Заболяването може да се появи при мъже и жени от всички възрасти.
Децата често развиват адипозогенитален синдром, а понякога и други признаци на ендокринна недостатъчност. Често се установяват промени в костната система: деформация на стъпалата, гръден кош, гръбначен стълб, дифузна остеопороза.
Отличителна чертаФормата на Дюшен е висока степенхиперензимията вече е при ранни стадииразвитие на процеса. По този начин нивото на ензим, специфичен за мускулната тъкан - креатинин фосфокиназа - в кръвния серум може да надвишава десетки и дори стотици пъти нормални показатели. Рязкото (10-100 пъти) увеличение на креатининфосфокиназата (CPK) при нервно-мускулна патология трябва да доведе до обсъждане предимно на следните заболявания: болест на Дюшен, болест на Бекер, полиомиозит и дерматомиозит, пароксизмална миоглобулинурия, дистална миодистрофия. Само в напреднал стадий на заболяването степента на хиперензимия постепенно намалява. Има съобщения за повишена СК на етапа на вътрематочно развитие.
Забавената мускулна релаксация е типична след мускулна треска. Последствията са мускулна слабост и ограничаване на движението на краката, ръцете и ръцете и влошаване на фината моторика. Някои болни мускули са особено силни поради типичния тъканен състав на подлежащите мускулни влакнавградени в мастна и съединителна тъкан.
Означава се още като гръбначен мозък, влияещ мускулен спазъм. Съществува до 30 различни формиспинална мускулна атрофия. Най-честата форма е проксималната спинална мускулна атрофия, описана тук. Означава се след началото на фюзелажа, както следва.
Мускулната дистрофия на Дюшен се предава по Х-свързан рецесивен начин. Генът е локализиран на късото рамо на X хромозомата. Честотата на генната мутация е доста висока (30%), което обяснява голям бройспорадични случаи.
Мутация (най-често делеция) води до полова или почти пълна липса на генния продукт - структурния протеин на дистрофията. Физиологична ролядистрофия не е напълно установена. Намира се във високи концентрации в областта на сарколемата, като очевидно играе определена роля в поддържането на целостта на тази мембрана. Липсата на дистрофични причини структурни променив сарколемата, което от своя страна води до загуба на вътреклетъчни компоненти и повишен приток на калций, което в крайна сметка води до смърт на миофибрите. Смята се, че дефицитът на дистрофия в синаптичните зони на кортикалните неврони е причина за умствена изостаналост.
Причината вероятно е генетичен дефект. Нервни клеткив гръбначния мозък клетките на предния рог са заразени. Смята се, че причината е нервната система от нерви. Това се отнася само за двигателната нервна система. Части от нервната система, които са отговорни за усещането за допир, болка и температура, остават непокътнати. функция Пикочен мехури ректума не се влошава.
Означението също така представлява загуба на мускули, причинена от нерви. Причината почти винаги е генетичен дефект, нервните обвивки стават необичайно дебели или самите нервни обвивки се влошават. На нервни влакназасяга ръцете и краката.
За медицинското генетично консултиране е много важно да се установи хетерозиготно носителство. С мускулна дистрофия на Дюшен при хетерозиготи, в приблизително 70% от случаите субклинична и понякога дори очевидни признацимускулна патология - известно удебеляване и дори уголемяване на мускулите на прасеца, бърза мускулна умора при физическа активност, промени в ЕМГ и при патоморфологично изследване на мускулни биопсии. Най-често хетерозиготните носители проявяват повишена активност на креатининфосфокиназата.
Интензивността на скоростта на нервната проводимост се забавя. Начало в долните крайници с мускулна загуба и свързана мускулна слабост. След това симптомите прогресират към долните крайници, като по-късно засягат ръцете и предмишниците. Сензорното увреждане е ниско. Възможни са автономни смущения, като твърде много или твърде малко производство на пот и нарушаване на основния кръвен поток. Могат да се появят леки симптоми на спазми в краката.
Мускулните слабости са резултат от мускулна слабост, докато мускулите са напълно функционални, което води до значителни, сериозни ограничения в движението на краката, ръцете и ръцете. Мускулите са много важна част от нашето тяло. Без мускули тялото губи способността си да се движи и да извършва различни дейности. Всъщност без човешки мускулна система, вероятно няма да можете да оцелеете. Това се дължи на факта, че повечето органи в храносмилателната системасе състои от мускули и дори самото сърце, което изпомпва кръв, също е мускул.
Ако има клинична картина на мускулна дистрофия на Дюшен при жени, първо трябва да се изключи възможността за аномалия на X хромозомата - синдром на Shereshevsky-Turner (TS), синдром на Morris (XY) или мозаицизъм за тези синдроми.
Мускулната дистрофия на Дюшен, която започва да се развива в пренаталния период, е по същество вродена миопатия и може да бъде диагностицирана скоро след раждането чрез извършване на мускулна биопсия и определяне на CPK активността.
Мускулна дистрофия на Бекер.Наред с тежката, злокачествена форма на Х-свързаната мускулна дистрофия на Дюшен, има и доброкачествена форма - болест на Бекер. По отношение на клиничните симптоми, тя е много подобна на формата на Дюшен, но като правило започва по-късно - на 10-15 години, протича леко, пациентите остават работоспособни дълго време, на възраст 20- 30 години и по-късно те все още могат да ходят. Плодовитостта не е намалена, така че понякога заболяването може да се проследи в няколко поколения на семейството: болен мъж чрез дъщеря си предава болестта на внука си („ефект на дядото“). Първоначалните симптоми, както при болестта на Дюшен, се проявяват като слабост в мускулите на тазовия пояс, след това в проксималните части на долните крайници. Походката на пациентите се променя, изпитват затруднения при изкачване на стълби или ставане от ниска седалка. Характерна е псевдохипертрофия на мускулите на прасеца. Ретракцията на петата (ахилесовите) сухожилия е по-слабо изразена, отколкото при болестта на Дюшен.
При тази форма няма интелектуални нарушения, кардиомиопатията липсва или е слабо изразена.
Както при други Х-свързани миодистрофии, при формата на Becker активността на CPK се увеличава значително, макар и в по-малка степен, отколкото при болестта на Дюшен, не надвишавайки 5000 единици. Генът за болестта на Бекер, подобно на болестта на Дюшен, е локализиран в късото рамо на X хромозомата; вероятно е и двата локуса да са тясно свързани или алелни. За разлика от болестта на Дюшен, при която практически няма дистрофия, при болестта на Бекер се синтезира анормална дистрофия. Разлики се откриват и при мускулна биопсия. При мускулната дистрофия на Бекер мускулните влакна обикновено са незакръглени, хиалинните влакна, характерни за мускулната дистрофия на Дюшен, се наблюдават изключително рядко.
Миодистрофия на Landouzy-Dejerine (фациоскапулохумерална миодистрофия).Заболяването се предава по автозомно-доминантен начин с висока пенетрантност, но донякъде променлива експресивност. Среща се много по-рядко от мускулната дистрофия на Дюшен (0,4 на 100 хил. население). Смята се, че генът за това заболяване е локализиран на хромозома 4. Жените боледуват по-често от мъжете (3:1), Физическо претоварване, интензивен спорт, както и нерационално физиотерапияможе да допринесе за по-тежкия ход на заболяването.
Мускулната дистрофия на Landouzy-Dejerine е сравнително благоприятна текуща форма на мускулна патология. Започва на около 20-годишна възраст, понякога по-късно. Въпреки това, при фамилни случаи на заболяването, когато е възможно да се проследят по-млади членове на семейството във времето, е възможно да се открие известна мускулна слабост, например мускулите на лицето, в по-ранна възраст.
Мускулна слабост и атрофия първо се появява в мускулите на лицето или раменния пояс. Постепенно тези нарушения се разпространяват към мускулите на проксималните ръце, а след това и към долните крайници. В повечето случаи първо се засягат мускулите на предната повърхност на краката (с развитието на спад на стъпалото), след това мускулите на проксималните крака. В разгара на заболяването кръговите мускули на окото и устата, големият гръден мускул, предният и долният трапецовидни мускули, широкият гръбен мускул, бицепсът, трицепсът на брахиите са силно засегнати. Характеристика външен видпациенти: типично лицемиопатия с "напречна усмивка" ("усмивка на Джоконда"), издатина Горна устна(„устни на тапир“), изразени крилообразни лопатки, особена деформация на гръдния кош с неговото сплескване в предно-задната посока и ротация навътре в раменните стави. Често има асиметрия на лезията, дори в рамките на един мускул (например мускул orbicularis oris). Може да се наблюдава псевдохипертрофия на прасеца, делтоидните мускули, а понякога и на лицевите мускули. Контрактурите и ретракциите са умерено изразени. Сухожилни рефлекси дълго времесе запазват, но понякога намаляват още в ранен стадий.
Рядко се откриват признаци на увреждане на сърдечния мускул. Серумната ензимна активност е леко повишена и може да бъде нормална. Интелигентността не страда. Продължителността на живота в повечето случаи не намалява. Интересно е, че ЕМГ при миодистрофия на Landouzy-Dejerine често не е напълно типична за мускулното ниво на лезията. При някои пациенти (членове на едно и също семейство) може да се наблюдава намаляване на амплитудата на биопотенциалите, тип крива на интерференция; при други, напротив, намаляване на честотата и хиперсинхронна активност, понякога с типичен палисаден ритъм. Човек трябва да е наясно със спиналния вариант, който имитира болестта на Landouzy-Dejerine.
Мускулна дистрофия на Erb-Roth (мускулна дистрофия на пояса на крайниците).Предава се по автозомно-рецесивен начин и двата пола са засегнати еднакво. Началото на заболяването в повечето случаи датира от средата на второто десетилетие от живота (14-16 години), но се описва като ранна форма на псевдо-Дюшен, когато първите симптоми се появяват преди 10-годишна възраст и заболяването е тежко, и късен вариант с начало след 30 години.
Протичането на заболяването може да бъде бързо или по-бавно, като средно пълната инвалидност настъпва 15-20 години от появата на първите симптоми. Миодистрофията започва или с увреждане на мускулите на тазовия пояс и проксималните крака (форма на Leiden-Moebius), или от раменния пояс (форма на Erb). В някои случаи раменният и тазовият пояс са засегнати едновременно. Мускулите на гърба и корема страдат доста значително. Болните имат характерна „патешка” походка, трудно стават от легнало или седнало положение, подчертано лумбална лордоза. В повечето случаи лицевите мускули не са засегнати. Контрактурите и псевдохипертрофиите са необичайни за тази форма. Може да настъпи крайна атрофия и ретракция на сухожилията. Интелигентността обикновено е запазена. Сърдечният мускул е почти незасегнат. Нивата на серумните ензими обикновено са повишени, но не толкова драстично, както при Х-свързаната мускулна дистрофия. Има индикации, че пациентите от мъжки пол имат по-високи нива на CPK от пациентите от женски пол. Има значителна разлика в експресията на мутантния ген между различните членове на семейството - заедно с тежката клинична картинаМоже да има относително леки и дори замъглени клинични симптоми. Смъртта обикновено настъпва от белодробни усложнения.
Тъй като клиниката на миодистрофията на пояса на крайниците е особено лесно имитирана от нервно-мускулни заболявания от различен характер, е необходимо, особено в спорадични случаи и с късно началозаболявания, провеждайте внимателно клиничен прегледза изключване на спинална амиотрофия, полимиозит, метаболитни, ендокринни, токсични, лекарствено индуцирани, карциноматозни миопатии. В миналото е имало ясна свръхдиагностика на тази форма на мускулна дистрофия.
Лечение на мускулни дистрофии.Терапевтичните възможности за мускулни дистрофии са много ограничени. Етиологични и патогенетично лечениепрактически не съществува. Симптоматичното лечение е насочено предимно към предотвратяване на развитието на контрактури, поддържане на съществуващата мускулна сила и евентуално леко намаляване на скоростта на развитие на атрофия. Основната задача е да се увеличи максимално периодът, през който пациентът може да се движи самостоятелно, тъй като контрактурите, сколиозата и респираторните нарушения бързо се увеличават в легнало положение. Медицински комплекстрябва да включва терапевтични упражнения, масаж, ортопедични мерки и лекарствена терапия.
Лечебната гимнастика се състои от пасивни и активни движения, извършвани във всички стави в различни позиции: изправени, седнали, легнали, с различно положение на крайниците. Активни движенияЗа предпочитане е да се изпълнява в изометричен режим. Часовете по гимнастика трябва да се провеждат редовно няколко пъти на ден. В същото време трябва да се внимава срещу прекомерни упражнения, особено когато са придружени от преразтягане на мускулите. Важни са дихателните упражнения (особено след обездвижване на пациента).
Ортопедични меркиконсервативни (специални шини) и хирургични (ахилотомия, трансекция на стомашно-чревния мускул), насочени към коригиране на контрактури и възникващи патологични изравнявания на крайниците, също са насочени към запазване на способността за самостоятелно движение. Във всеки случай е необходимо индивидуално да се претеглят очакваните ползи и възможна вредаот операция. Трябва да се има предвид, че често (по-специално при тежка хиперлордоза и слабост на мускула на четириглавия бедрен мускул) еквиноварусната позиция на краката има компенсаторна стойност и след, например, ахилотомия, пациентът може да бъде напълно обездвижен. При развитие на контрактури се препоръчва леко разтягане на мускулите до 20-30 пъти на ден, последвано от поставяне на шина по време на сън.
Лекарствената терапия включва предписване на метаболитни лекарства, насочени към попълване на дефицита на енергия и протеини, но тяхната ефективност е много съмнителна. Използват се калциеви антагонисти (поради дефекта, идентифициран при болестта на Дюшен клетъчни мембрани, което води до повишено навлизане на калций в клетката), имуномодулатори, фосфорсъдържащи съединения (АТФ, фосфаден), витамин Е (100 mg перорално 3 пъти на ден). Доказано е, че при болестта на Дюшен употребата на преднизолон (0,75 mg / kg на ден) може драстично да увеличи мускулната сила, но този ефект продължава не повече от година и като цяло не влияе върху изхода на заболяването. Поради сериозни странични ефекти, възникващи лъжи продължителна употребалекарство, употребата му е неподходяща. Оценки на ефекта анаболни стероидиса противоречиви и целта им често е свързана с неоправдан риск. При оценката на ефекта на някои лекарства при болестта на Дюшен трябва да се има предвид, че при умерена тежест на заболяването при пациенти на възраст 3-6 години може да има относително стабилизиране на състоянието, свързано с възрастовото развитие на мускулите система, придобиване на двигателни умения, които могат до известна степен временно да компенсират непрекъснато протичащия дегенеративен процес.
Коригирането на диетата на пациента е от известно значение, препоръчва се диета с високо съдържание на протеини, ниско съдържание на мазнини и ниско съдържание на калории с оптимално съдържание на витамини и микроелементи. Психологическата подкрепа за пациента, продължаващото обучение и правилното професионално насочване играят важна роля.
Страница 44 от 44
Скелетните мускули участват в патологичен процесза различни дегенеративни, метаболитни и възпалителни заболявания. В повечето случаи това причинява дегенерация на мускулните влакна, а при хронични форми - тяхното заместване съединителната тъкани мазнини. Проксималните мускулни групи са по-значително увредени от дисталните, както и долните крайници спрямо горните. Болното дете има така наречената патешка (клатеща се) походка и не може да тича, да се катери по стълби или да се изправя, ако е в седнало положение. Неговите сухожилни рефлекси са потиснати, степента на тяхното изчезване е пропорционална на степента на отслабване на мускулната сила. Чувствителността не е нарушена.
Диагностично ценните лабораторни методи включват определяне на активността на ензимите, особено креатинфосфокиназата, в серума. Този ензим, който катализира реакцията: фосфокреатин + АДФ-креатин + АТФ, присъства главно в мозъчните клетки и мускулната тъкан. При някои дифузни мускулни заболявания, особено при мускулна дистрофия, излишните му количества проникват в междуклетъчното пространство и кръвта. Пациентите обикновено имат повишена активност на серумната лактат дехидрогеназа и глутаминова оксалоцетна трансаминаза, но широкото им разпространение в други тъкани, включително черния дроб, намалява специфичността на теста. Обикновено за изясняване на диагнозата е необходима биопсия на мускулна тъкан.
Възпалителни мускулни заболявания. Възпалението на мускулната тъкан придружава някои инфекции, особено трихинелоза, токсоплазмоза и тези, причинени от вируса Coxsackie. Често е компонент на колагенови заболявания, включително дерматомиозит, лупус еритематозус, нодозен периартериит и ревматоиден артрит.
Полимиозит. Дифузно изолирано мускулно възпаление с неизвестна етиология се нарича полимиозит. Характеризира се с бързо прогресиращ ход, слабост и болка в проксималните мускулни групи. Често в процеса участват мускулите на врата, което затруднява детето да повдигне главата си и да я задържи в това положение. Лабораторните признаци на мускулно възпаление включват повишаване на ESR и броя на белите кръвни клетки. Липсата им обаче не изключва полимиозит. Нивата на серумните ензими обикновено са повишени. При мускулна биопсия се определят дегенерация и частична регенерация на влакна и тяхната инфилтрация от лимфоидни клетки. Трудно е да се разграничи полимиозит от мускулна дистрофия и дерматомиозит. Може да представлява атипична форма на дерматомиозит, въпреки че хистологичната картина на двете състояния е малко по-различна: дерматомиозитът се характеризира с васкулит, който обикновено липсва при полимиозит. Прогнозата за последното е малко по-благоприятна. Лечението с кортикостероиди е съпроводено с ефект, но при спирането им може да настъпи рецидив.
Прогресивен осифициращ миозит. Етиологията на това рядко заболяване на съединителната тъкан и мускулите е неизвестна. Съобщава се, че засяга братя и сестри, включително близнаци, и се предава на кръвни роднини по права линия. Предполага се, че заболяването се унаследява по автозомно-доминантен начин. Момчетата боледуват 2-3 пъти по-често от момичетата.
Патологичните признаци зависят от стадия на заболяването. В ранните стадии се открива локален оток и възпалителни клетъчни инфилтрати в мускулите и сухожилията. По-късно зоните на възпаление се заместват от гранулационна тъкан и в крайна сметка в лезиите се образуват области от хрущялна и костна тъкан.
Почти 75% от болните деца са диагностицирани с рожденни дефектиразвитие, най-често недоразвитие на пръстите и анкилоза на фалангите на първите пръсти и недоразвитие на първите пръсти, полидактилия, изкривяване на пръстите, синдактилия (крака), деформация уши, глухота, липса на зъби. Същите вродени дефекти могат да присъстват и при роднини на пациента, които не са развили прогресивно заболяване на съединителната тъкан и мускулите. Възрастта, на която може да започне осифициращият миозит, варира от раждането до по-голяма детска възраст. Обикновено има три етапа: 1) на местата на малки локални наранявания се появяват ограничени, често топли и меки на допир тестообразни отоци на меките тъкани; 2) след няколко дни симптомите на възпаление изчезват и лезията се втвърдява; 3) настъпва осификация на засегнатата област. Периодично се появяват нови лезии, главно в областта на шията и гърба. Основен симптомТортиколис може да се развие, ако процесът се е развил в стерноклеидомастоидния мускул. Осификацията в крайна сметка се разпространява до много сухожилия и връзки. Появява се анкилоза на гръбначния стълб и ставите на ръцете и краката (фиг. 21-5). Възпалението може да се разпространи до темпорамандибуларните стави, затруднявайки дъвкателните движения. Костните шипове могат да изпъкнат през кожата. В юношеска възраст заболяването често води до пълна неподвижност и смърт поради дихателна недостатъчности спиране на дишането, въпреки че има съобщения за оцеляване. При осифициращ миозит има висок риск от развитие на остеогенен сарком.
Ориз. 21-5. Дете с прогресиращ осифициращ миозит (типична поза със скованост на мускулите на врата и гърба).
Понякога патологичният процес е ограничен до мястото на предишно увреждане на меките тъкани (miositis ossificans circumscripta). Широко разпространена калцификация на мускулната тъкан може да възникне и при хроничен полимиозит и дерматомиозит.
резултати лабораторни методиизследванията нямат диагностична стойност.
Серумните нива на калций, фосфор, алкална фосфатаза и активността на креатин фосфокиназата и други ензими остават нормални. Костенпри източника на увреждане не се различава по структура от нормата.
Съществуващи методилеченията са незадоволителни. В някои случаи се наблюдава забавяне на прогресията на заболяването при употребата на ACTH и други кортикостероиди. Тяхната роля за крайния резултат от лечението е под въпрос.
Ендокринни и метаболитни миопатии. Миопатията при хипертиреоидизъм е доста рядко усложнение. Характеризира се с птоза, двустранна пареза на лицевите мускули и мускулите на проксималните крайници. В този случай някои симптоми на хипертиреоидизъм могат да бъдат маскирани от мускулна слабост, но тахикардия, повишено изпотяване и повишено щитовидната жлеза. Сухожилните рефлекси, за разлика от много други форми на миопатия, остават нормални. След корекция на хипертиреоидизма мускулната слабост постепенно изчезва.
Миопатия при хипотиреоидизъм. Хипотиреоидизмът при кърмачета може да бъде свързан с мускулна слабост и хипотония. При по-големи деца с микседем мускулните контракции и релаксации се забавят, а в някои случаи се наблюдава мускулна хипертрофия (синдром на Debreu-Semelen). Комбинацията от признаци като слабост и мускулна хипертрофия предполага мускулна дистрофия.
Миопатия по време на лечение с кортикостероиди. Може да усложни болестта на Иценко-Кушинг, но по-често се развива при лечение с големи дози синтетични стероиди. Слабостта е особено забележима в мускулите на тазовия пояс, което се проявява в клатеща се (като патешка) походка, затруднено изкачване на стълби и опит за изправяне от седнало положение. Няма рефлекс на коляното. Може да настъпи изтъняване на мускулите. Миопатичните промени в мускулната тъкан обикновено са незначителни дори при тежка слабост. Мускулна силаслед спиране на кортикостероидите възстановяването е бавно (за няколко месеца).
Миопатия при хиперпаратироидизъм. Хиперпаратироидизмът може да бъде свързан със слабост и хипорефлексия, причинени от хиперкалиемия. Те обикновено изчезват бързо след паратироидектомия.
Дефицитът на карнитин (липидна миопатия) е съпроводен с натрупване на големи количества липиди в мускулите и в резултат на това нарушаване на енергийното им захранване. Карнитинът е основен компонент на системата, която осигурява преноса на мастни киселини от дълга веригаот цитозола до митохондриите, където претърпяват окисление. Мускулна слабост се развива при две форми на дефицит на карнитин.
Дефицитът на карнитин в мускулите се проявява клинично с прогресираща слабост на техните проксимални групи, по-често при ученици и юноши. Понякога слабостта е периодична и се комбинира с миоглобинурия. В тежки случаи може да настъпи парализа на дихателните мускули. Нивата на серумните ензими (креатинкиназа и алдолаза) се повишават. Електромиограмата разкрива неспецифични промени, характерни за миопатия. В мускулната биопсия можете да видите голям брой мастни капчици. Серумните нива на карнитин не се променят, но нивата на мускулния карнитин намаляват. Разпознаването на патологията е от съществено значение, тъй като тя може да бъде лечима. Често се бърка с мускулна дистрофия. Ефектът може да настъпи след перорално приложение на 100 mg/(kg/ден) карнитин. В някои случаи лечението с кортикостероиди е ефективно.
- Системният дефицит на карнитин се проявява чрез прогресивна миопатия, включително кардиомиопатия и чернодробна дисфункция, придружена от клинична картина на чернодробна енцефалопатия от типа на синдрома на Reye. Дефицитът на карнитин се различава от последния с повтарящия се курс и тежка мускулна слабост, която продължава между периодите на обостряне на енцефалопатията. Нивата на серумната креатинфосфокиназа са значително повишени, а нивата на карнитин са намалени както в серума, така и в мускулите. Промените в биопсичния образец са подобни на тези при дефицит на карнитин в мускулната тъкан. Подобни клинични и морфологични промени, включително дефицит на карнитин, могат да бъдат открити при нарушения на метаболизма на органичните киселини, например при метилмалонова и глутарова ацидурия (вторичен дефицит на карнитин).
Ориз. 21-6. Дете с вродена липса на ляв голям гръден мускул. 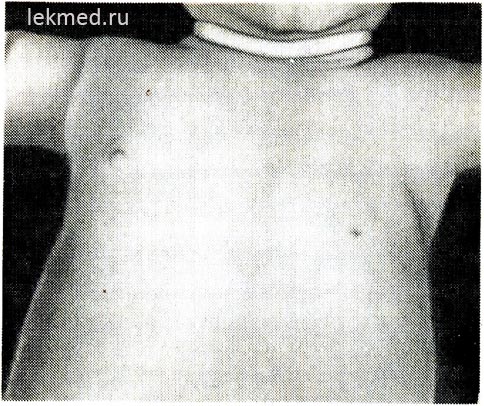
Трябва да се отбележи липсата на предната аксиларна гънка и ниско разположеното зърно.
Лечението се състои в това пациентът да следва диета, богата на въглехидрати и бедна на мазнини, и да приема карнитин в дневна доза от 100 mg/kg.
Вродени мускулни дефекти. Вродена липса на мускул. Мускулното недоразвитие може да бъде доста често срещано и да доведе до пълно блокиране на ставните движения или вродена артрогрипоза. Като вроден дефект най-често липсва един мускул. Доста често срещана аномалия е липсата на стерналната част на големия гръден мускул (фиг. 21-6), в някои случаи този дефект се комбинира със синдактилия от засегнатата страна (синдром на Полша). Липсата на гръдния мускул често придружава мускулна дистрофия. Вродената липса на коремни коремни мускули често се свързва с дефекти в развитието на пикочните пътища.
Ориз. 21-7. Деформация на врата и асиметрия на лицето при момче с вроден тортиколис, нелекуван от 12-годишна възраст. 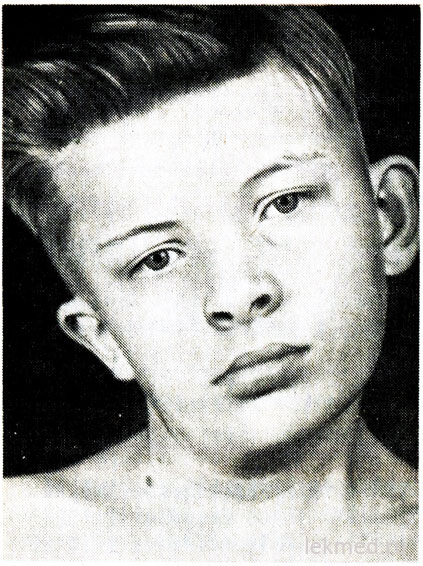
Вроденият тортиколис се причинява от едностранно скъсяване или контрактура на стерноклеидомастоидния мускул. Главата на пациента е наклонена към контрактурата, а брадичката е насочена надолу в обратна посока (фиг. 21-7). При опит за коригиране на позицията на главата се усеща значително мускулно съпротивление. В засегнатия мускул се усещат области на уплътняване. Причината за дефекта е неясна; дълго време се смяташе, че е резултат от родова травма. Въпреки това, тортиколис се среща при деца, родени чрез операция цезарово сечение; това предполага, че в някои случаи причината за дефекта е свързана с пренаталния период. Тортиколисът трябва да се диференцира от патологичен наклон на главата поради деформация на шийните прешлени, например с аномалия на Klippel-Weil, и от фрактури или дислокации на шийните прешлени. Те се изключват чрез радиографско изследване. При по-големи деца може да се появи накланяне на главата при страбизъм, дистония, тумори на задната черепна ямка и шийни прешленигръбначен мозък, осифициращ миозит, цервикален лимфаденит или диафрагмална херния. В повечето случаи вроденият тортиколис може да бъде коригиран с терапевтични упражнения. Въпреки това, когато хронична форматортиколис води до асиметрично развитие на лицето и главата (виж Фигура 21-7), което може да наложи мускулна дисекция по козметични причини.
Вродени миопатии. Тази група включва няколко редки форми на наследствени заболявания, при които мускулна слабост и хипотония се появяват от ранна детска възраст (виж Таблица 22-1). Тяхната точна диагноза има голямо значениеот гледна точка на прогнозата. Като цяло е благоприятен за нормалното функциониране и продължителността на живота, за разлика от болестта на Вердниг-Хофман или вродената мускулна дистрофия. Откриването на вродени миопатии обикновено се улеснява чрез мускулна биопсия.
- Заболяване на централното ядро. Централната част на мускулните влакна е необичайно оцветена, но равномерно. Електронномикроскопското изследване разкрива намаляване на броя на митохондриите и изчерпване на саркоплазмения ретикулум в централната част на влакната.
Немалинова миопатия. Терминът "немалин" се обяснява с факта, че в мускулните влакна са дефинирани нишковидни структури.
Ориз. 21-8. Миотонична контракция на езика (а) с рязък удар с перкусионно чукче върху дясната му половина и клепачите (б) при дете с хиперкалиемична форма на фамилна периодична парализа. 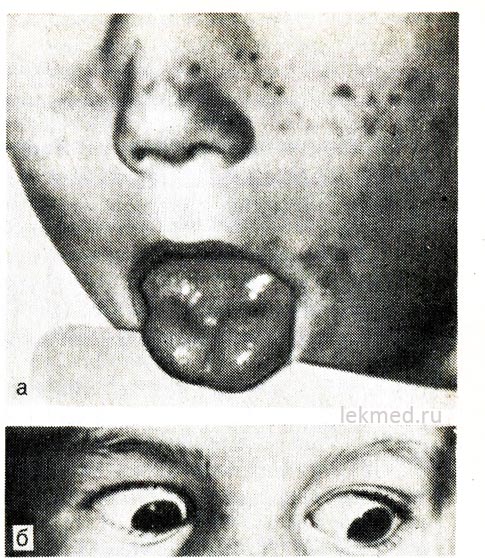
При гледане надолу клепачът остава свит.
Електронномикроскопските данни показват, че това е резултат от промени в Z-лентите на миофибрилите.
Митохондриални миопатии. Има съобщения за някои форми на миопатия, при които най-важните промени настъпват в митохондриите на мускулните влакна. Те могат значително да се увеличат както по брой, така и по размер. Мускулната слабост и хипотонията могат да бъдат открити още в ранна детска възраст, но понякога те прогресират забележимо едва в училищна възраст. Кардиомиопатия, енцефалопатия и лактатна ацидемия често придружават миопатиите от тази група.
Миотония. Това състояние е признак на различни мускулни заболявания, като дистрофична миотония, хиперкалиемична форма на фамилна пароксизмална парализа и заболявания на съхранението на гликоген. Миотонията се определя като значително забавяне на мускулната релаксация след доброволни или принудителни контракции. Клинично се проявява в невъзможност за свиване на юмрука или във видимо продължително свиване на мускулите след тяхното дразнене, изразяващо се в рязко дразнене (фиг. 21-8). Това може да се наблюдава, ако ударите повърхностна мускулна група с перкусионен чук, например мускулите на езика или палмарната повърхност в областта на издигането на първия пръст. Миотонията се потвърждава от данните от електромиографията. В този случай се забелязва характерна спонтанна мускулна активност след релаксация или доброволно свиване (миотоничен разряд).
Вродена миотония (болест на Томсен). Единственият признак на това заболяване, унаследен по доминантен тип, е миотонията. Може да се прояви в ранна детска възраст под формата на забавяне на преглъщането и последващо повръщане.
резултат от невъзможност за нормално отпускане на мускулите на фаринкса. При по-стари детствомиотонията се проявява като неспособност на пациента да разхлаби пръстите си, стиснати в юмрук. При първия опит да се направи някакво движение, мускулите на детето стават твърди. При многократно повтаряне на едно и също движение те донякъде се отпускат. Например, болно дете изпитва голяма трудност да започне да ходи. Обикновено прави първите няколко крачки много колебливо и бавно. След няколко секунди походката става нормална или почти нормална. Симптомите на миотония се влошават от неблагоприятно емоционално състояние на пациента и охлаждане на тялото. Мускулната сила остава нормална, мускулите са достатъчно развити и често забележимо уголемени, което създава погрешно впечатление за атлетичната конструкция на пациента.
Диагнозата се основава на клинични данни и електромиографски данни. Серумната ензимна активност е в нормални граници. Единственият хистологичен признак е хипертрофия на мускулни влакна.
Заболяването се различава от дистрофичната миотония по липсата на мускулна слабост и атрофия и дистрофични промени в биопсията на мускулната тъкан. Лечението с новокаин или хинидин сулфат е придружено от ефект и е показано при функционални нарушения. Протичането на заболяването обикновено е доброкачествено и състоянието на пациента може да се подобри с възрастта.
Пароксизмална парализа.Тази група заболявания се характеризира с периодична мускулна слабост с пълно или почти пълно възстановяване на мускулната сила в периода между атаките. Това включва и дефицит на мускулна фосфорилаза (болест на McArdle).
Хиперкалиемична пароксизмална парализа.Наследствената епизодична адинамия или парамиотония се предава по доминантен начин и е особено тежка при мъжете. Обикновено започва в ранна детска възраст (понякога в ранна детска възраст). Атаките възникват по време на почивка след тежко мускулно натоварване. Слабостта се развива бързо и може да продължи няколко часа. Особено се усеща в краката; дихателната функция обикновено не е нарушена. Често адинамията е придружена от миотония, която продължава между атаките, което се проявява най-ясно под формата на забавяне на движението на клепачите при гледане надолу (виж Фиг. 21-8, b).
Нивата на серумния калий често са повишени по време на атака, но може да се наложи повторно тестване при множество атаки, за да се определи това надеждно. Пристъпът може да бъде изкуствено провокиран чрез натоварване с калий (2-3 g перорално), но трябва да се извършва само под ЕКГ мониториране. Повтарящите се атаки се спират с диакарб. Тежките форми на заболяването се характеризират с развитие на хронична, лека слабост и дистрофични промени в мускулите.
Хипокалиемична пароксизмална парализа. Фамилната пароксизмална парализа, също унаследена по доминантен начин, е особено тежка при момчетата. За разлика от хиперкалиемичната форма, първият пристъп се появява в късна детска или ранна юношеска възраст. Причината е консумацията на обилни ястия, богати на въглехидрати, или почивка след това физическа дейност. Обикновено пристъпът започва при следващата сутринслед тежко физическо натоварване и обилна вечеря. Характеризира се с мускулна слабост и арефлексия. Дихателната функция може да бъде нарушена. Може да се появи аритмия, включително камерни екстрасистоли и тахикардия. Пристъпите могат да продължат повече от 24 ч. В паралитичната фаза нивото на калий в серума обикновено намалява (2-3 mmol/l). Основният дефект е неизвестен. Пациентите с повтарящи се тежки пристъпи развиват хронична мускулна слабост и патологични променив мускулите. Лечението по време на пристъпи се състои в приемане на калиев хлорид; началната му доза е 2-3 г. Diacarb помага за намаляване на честотата на атаките.
Пароксизмална миоглобинурия (идиопатична миоглобинурия). Идиопатичната миоглобинурия е хетерогенна група от заболявания, при които пристъпите на парализа с миоглобинурия възникват спонтанно или след интензивно физическо натоварване. Заболяването се унаследява по доминантен начин, свързано с Х-хромозомата. Мускулите, най-често мускулите на прасеца и бедрата, стават болезнени и подути по време на пристъп. Урината става тъмночервена или кафяв цвят. Миоглобинурията може да причини некроза на бъбречните тубули, което води до смърт бъбречна недостатъчност.
Диагнозата се потвърждава от откриването на миоглобулин в урината. Положителният тест за бензидин при липса на червени кръвни клетки в урината потвърждава наличието на миоглобин в него, особено ако хемоглобинът не се открива в серума. Хемоглобинът се определя чрез спектрофотометрия. Пароксизмалната миоглобинурия трябва да се разграничава от болестта на McArdle, дефицита на карнитин палмитил трансфераза и миоглобинурията след необичайна интензивна физическа активност или мускулна травма в здрав човек. Миоглобинурия след тежко мускулно натоварване възниква при псевдохипертрофична мускулна дистрофия (болест на Дюшен).
Лечението се състои в почивка на легло; ако е необходимо, изпълнете изкуствена вентилациябели дробове. За да се предотврати бъбречна недостатъчност, е необходимо да се предпише на пациента много течности.
Дефицит на карнитин палмитил трансфераза.Когато този ензим е дефицитен, прехвърлянето на дълговерижни мастни киселини към митохондриалните сегменти, в които протича окисление и производство на кетони, е нарушено. Изоензимният дефицит тип II се унаследява по рецесивен начин. Поради неговия дефицит се нарушава кетогенезата в тъканите, включително мускулите и черния дроб. Първите признаци на заболяването се появяват по-често при деца в училищна възраст и юноши. Те се състоят от повтарящи се епизоди на мускулна болка, слабост и треска след тренировка или гладуване. Миоглобинурията, придружаваща атаките, може да доведе до бъбречна недостатъчност. Гладуването води до хипогликемия. Между атаките децата изглеждат здрави. Заболяването трябва да се диференцира от други състояния, придружени от периодична слабост и миоглобинурия. Методът за определяне на активността на карнитин палмитил трансферазата има диференциално диагностична стойност. Намалява се в мускулите и чернодробна тъкан, левкоцити и фибробластна култура. Спазването на диета, състояща се от храни, обогатени с въглехидрати и ниско съдържание на мазнини, помага за намаляване на броя на атаките.
Мускулни дистрофии. Тези аномалии принадлежат към група фамилни заболявания, придружени от дегенерация на мускулни влакна. Класификацията на мускулните дистрофии се основава на характеристики като време на поява, скорост на прогресиране, разпределение на лезиите в мускулните групи и начин на унаследяване.
Псевдохипертрофична мускулна дистрофия. Детската, или Дюшен, е най-честата форма на мускулна дистрофия; честотата му е 0,14 на 1000 деца. В класическата си форма се среща само при момчета, а Х-свързаното унаследяване се среща при приблизително 50% от пробандите. В други случаи болестта се причинява от нови мутации. Съобщава се за рядка форма на мускулна дистрофия, клинично идентична с формата на Дюшен, но се унаследява по рецесивен начин с еднаква честота на заболяването при момчета и момичета. Рядко е възможно надеждно да се диагностицира заболяването при дете под 3-годишна възраст. Анамнезата обикновено показва, че детето е имало изоставане в развитието двигателни функции, той започна да седи, ходи и тича до късно, което, естествено, показва повече ранен стартзаболявания. Клатушкаща се (патешка) походка, затруднено изкачване на стълби, хипертрофия на мускулите на прасеца са чести клинични проявления. В някои случаи в процеса участват и други мускули, по-специално делтоидните, брахиорадиалните и езиковите мускули.
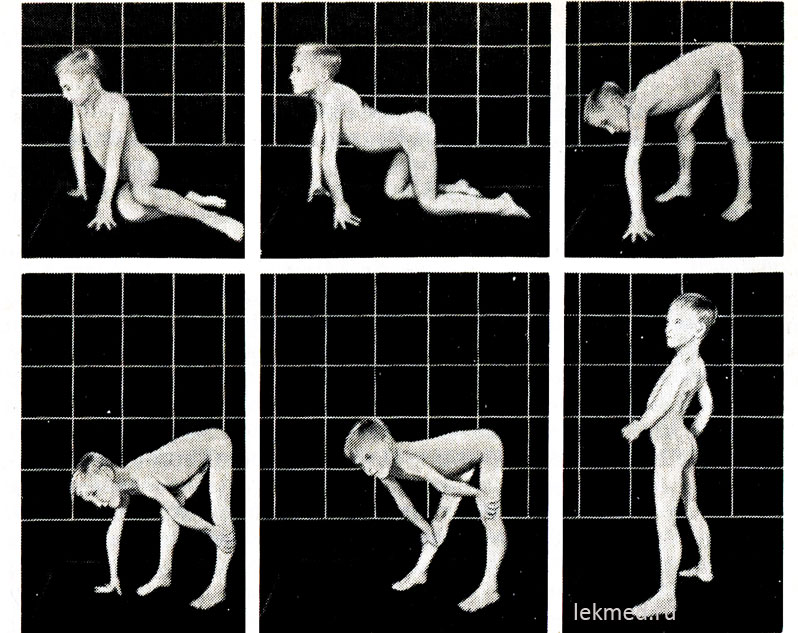
Ориз. 21-9. Типични пози, заети при изправяне от пода (знак на Gowers) от 7-годишно дете с псевдохипертрофична миопатия.
В изправено положение (последната снимка) има значително изразена лордоза.
В началото на заболяването хипертрофираните мускули имат значителна сила, но по-късно тя намалява (псевдохипертрофия), тъй като увеличаването на мускулната маса се дължи на тяхната мастна инфилтрация. Силата на хипертрофиралия мускул на прасеца значително надвишава силата на мускулите на предната повърхност на крака, което обяснява честите контрактури на сухожилието на петата и ходенето на детето на пръсти. Слабостта на мускулите на тазовия пояс се изразява в характерна патешка (лордска) походка и затруднения, които детето изпитва при изправяне от седнало положение на пода. При доста тежки форми на мускулна дистрофия детето проявява симптом на Говърс: когато става от пода, той първо коленичи, опирайки се на ръцете си, а след това се издига, последователно изтласквайки ръцете си от пищялите си, коленни ставии бедрата (фиг. 21-9). Слабостта на мускулите на раменния пояс може да се определи, като държите детето в повдигнато положение за подмишниците. Обикновено той се опитва да се задържи, като притиска ръце към тялото си; с мускулна дистрофия изглежда, че се изплъзва през ръцете на проверяващия. Болното дете често не може да вдигне ръце над главата си. В по-късните стадии на заболяването се развива значителна мускулна атрофия. Обикновено до 12-годишна възраст детето вече не може да ходи. Пациентите в 75% от случаите умират преди 20-годишна възраст. Повечето от тях имат кардиомиопатия, която в някои случаи причинява внезапна смърт. Ако наследството е свързано с Х-хромозомата и заболяването е започнало в късна детска възраст, продължителността на живота остава дълга (мускулна дистрофия на Бекер). Средният коефициент на интелигентност за деца с Дюшен е 80; 25% от децата имат умствена изостаналост.
При диференциална диагнозаМускулната дистрофия на Дюшен трябва да се вземе предвид болестта на Вердниг-Хофман при по-големи бебета и мускулни заболявания като ендокринни миопатии, дефицит на карнитин, заболявания на съхранението на гликоген и полимиозит. Понякога при контрактури на сухожилието на петата и ходене на пръсти на детето може да се предположи церебрална парализа, но при мускулна дистрофия няма характерни признаци. церебрална парализаспастичност и хиперрефлексия.
Диагнозата се основава на определяне на серумната ензимна активност, електромиографски данни и биопсия на мускулна тъкан. Ензимна активност, особено креатин фосфокиназа, още преди развитие клинични симптомичесто надвишава нормата 10 пъти дори при кърмачета. Електромиограмата показва предимно намаляване на продължителността и намаляване на амплитудата на двигателните потенциали. Хистологичните промени се състоят в дегенерация на мускулни влакна. Те често варират по размер и са частично заменени от мастна и съединителна тъкан. Размерът на техните ядра също варира. Диагнозата може да се постави при раждането чрез измерване на активността на креатинфосфокиназата. Методите за идентифициране на женски носители все още не са разработени, въпреки факта, че 60-80% от тях имат леко или умерено увеличение на нивото му. Тези признаци са по-типични за детството, отколкото за следващите периоди от живота.
Ефективни методиняма лечение. Пациентът трябва да бъде поддържан активен и да може да ходи колкото е възможно повече. Необходимо е да се гарантира, че детето избягва интензивна физическа активност, тъй като може да причини разкъсване на мускулните влакна. В някои случаи хирургичното удължаване на сухожилието на петата подобрява способността за ходене, но продължително почивка на леглослед ортопедична корекция може да се увеличи мускулна атрофия. Генетичното консултиране играе важна роля.
Вродена мускулна дистрофия. Заболяването се унаследява по автозомно-рецесивен начин и се характеризира с мускулна хипотония и слабост при кърмачета. То е включено в групата състояния, дефинирани като „отпуснато дете” (виж Таблица 21-1). Началото на заболяването датира от пренаталния период. Понякога новороденото има тежка мускулна атрофия, контрактури и ограничена подвижност на ставите. Разграничаването от болестта на Вердниг-Хофман е трудно. Фасцикулациите на езика, характерни за последния, липсват при мускулна дистрофия. Сухожилните рефлекси са потиснати, но не напълно загубени. Процесът включва мускулите, участващи в дишането, включително диафрагмата. В тежки случаи смъртта настъпва преди навършване на 1 година поради дихателна недостатъчност; при по-леките форми нормалната жизнеспособност се запазва дълго време. Не се отбелязва повишаване на активността на серумните ензими, въпреки че настъпват дистрофични промени в мускулите.
Лицево-хумерална форма на мускулна дистрофия. Този е достатъчен лека формаМускулната дистрофия се унаследява по автозомно-доминантен начин. Обикновено започва на възраст 10-20 години и се характеризира със слабост и атрофия на мускулите на лицето и раменния пояс. Лицето е напълно дружелюбно, пациентът не може да затвори очи и да свири. Заболяването прогресира бавно и е съвместимо с нормалната продължителност на живота. Диагнозата се основава на клиничните данни и начина на унаследяване. Резултатите от биопсия на мускулна тъкан показват дистрофични промени в нея. Нивата на серумната креатинфосфокиназа могат да останат в нормални граници или да бъдат леко повишени.
Тазова форма на мускулна дистрофия. Тази група хетерогенни заболявания се характеризира с бавна прогресия на мускулна дистрофия и се унаследява по автозомно-рецесивен начин. Началото на заболяването се отнася до по-старо детство, юношество или зряла възраст. Обикновено се засягат мускулите на тазовия пояс.
Очна форма на миопатия. Дистрофичните промени настъпват предимно във външните очни мускули. Заболяването започва в детството или юношеството. При него прогресира птозата и ограничаването на движенията очни ябълки. Понякога слабостта се разпространява към мускулите на лицето и шията. Заболяването трябва да се диференцира от миастения гравис и парализа черепномозъчни нервиза тумори на мозъчния ствол.
Прогресивната офталмоплегия, започваща в детството или юношеството, е свързана с атипична пигментна дегенерация на ретината и сърдечен блок (синдром на Kearns-Sayers). Обикновено се свързва с прогресивна атаксия, забавяне на растежа и пубертета. Под мускулната сарколема се откриват големи натрупвания на атипични митохондрии. Генетичният характер на този процес не е установен. Възможността за внезапна смърт от проблеми със сърдечната проводимост може да се контролира чрез използване на пейсмейкър.
Миотонична дистрофия. Въпреки факта, че миотоничната дистрофия започва като при възрастен, нейното начало все повече се записва при кърмачета и по-късна детска възраст. Унаследява се по автозомно-доминантен начин. Началото му в детска възраст показва, че майката страда от миотония. Съответно вътрематочните фактори могат да повлияят на тежестта на заболяването при детето. Още в момента на раждането той може да бъде определен мускулна хипотония, липсва му способността да суче. Забавеното физическо и умствено развитие обикновено се открива по-късно. В ранна детска възраст мускулната слабост и атрофия се разпространяват главно в лицевите, челюстните и темпоралните мускули. Обикновено се отбелязва двустранна птоза. Диагностично значимите методи включват мускулна перкусия, електромиография; типично за тези пациенти е невъзможността да се разхлаби ръката, стисната в юмрук (виж Вродена миотония). Слабостта и атрофията на мускулите на крайниците и тазовия пояс (обикновено дисталните групи) се откриват в по-стара детска или юношеска възраст. При възрастни това заболяване е придружено от катаракта, плешивост и атрофия на тестисите.
Диагнозата се основава на идентифициране на признаци на миотония, характерно разпределение на мускулна слабост, доминантно наследство и дистрофични промени в мускулите. В детска възраст ходът на заболяването може да бъде неблагоприятен и често се придружава от умствена изостаналост. До юношеството мускулната слабост излиза на преден план. При функционални нарушения е показано лечение с новокаин и хинидин.

