NERVU SISTĒMAS IESPĒJAMĀS SLIMĪBAS
16. LEKCIJA
Deģeneratīvas slimības, kas galvenokārt skar neiromuskulāro sistēmu, veido lielāko visu iedzimto slimību grupu.
Ārkārtīgi nozīmīgi un bieži vien izšķiroši neiromuskulāro slimību diagnostikā ir rezultāti elektrofizioloģiskās un bioķīmiskie pētījumi. Tikpat liela ir patomorfoloģisko atradumu nozīme. Muskuļu biopsijas materiāla pārbaude gaismas mikroskopā palīdz atšķirt miogēno atrofiju no neirogēnas atrofijas. Histoķīmiskā izmeklēšana ir nepieciešama, lai identificētu vielmaiņas muskuļu bojājumus, un elektronu mikroskopija atklāja veselu lielu slimību klasi – neprogresējošas miopātijas.
Maiga skarto locītavu stiepšana samazina kontraktūru rašanos un tādējādi izraisa lēnāku slimības progresēšanu. Šajā gadījumā tiek veikti dažādi specifiski vingrinājumi, un ir konstatēts, ka Urias spiedošais pārsējs ir efektīvs ekstremitāšu dziļuma traucējumu gadījumos un bieži tiek uzskatīts par izdalījumu ārstēšanu mājās. Sensomotoriskie treniņi pacientiem ar muskuļu traucējumiem tiek nodrošināti individuālā vai grupu terapijā un vērsti uz koordināciju un smalko motoriku.
Ļoti svarīga metodeĀrstēšana cilvēkiem ar muskuļu slimībām ir atjaunot spēju vadīt transportlīdzekli. Šim nolūkam notiek sadarbība ar ārējo autoskolu darba terapijas nodaļā, un braukšana Bādvildungenā notiek ar piemērotu invalīdu transportlīdzekli ar apmācītu braukšanas instruktoru.
Progresējoša muskuļu distrofija. Termins muskuļu distrofijas attiecas uz ģenētiski noteiktu traucējumu grupu, kam raksturīgi progresējoši deģeneratīvas izmaiņas muskuļu šķiedrās bez primārā perifērā (apakšējā) motorā neirona patoloģijas.
Dažādās formas atšķiras viena no otras pēc mantojuma veidiem, procesa sākuma laika, tā norises rakstura un ātruma, unikālās muskuļu atrofijas topogrāfijas, pseidohipertrofijas un cīpslu ievilkšanas esamības vai neesamības un citām īpašībām. .
Papildu pieeja muskuļu traucējumu rehabilitācijai ir adekvātas konsultācijas. Tas attiecas ne tikai uz ortozēm, bet arī uz visām pārējām palīglīdzekļi mājās, Mērķtiecīga palīdzība invalīdiem ar muskuļu traucējumiem ir ārkārtīgi izdevīga, ratiņkrēsli ir precīzi izmērīti un pielāgoti cilvēku ar muskuļu traucējumiem vajadzībām. Dušas krēsls, vannas lifts, ratiņkrēsli, rampas priekš ratiņkrēsli un citi.
Fizikālās terapijas nodaļā tiek izmantotas dažādas procedūras pacienta relaksācijai. Turklāt šī ārstēšanas nozare var mazināt bieži vien nelielus sāpju simptomus pacientiem ar muskuļu traucējumiem. Logopēdija ir vēl viena rehabilitācijas nozare muskuļu traucējumu ārstēšanai. Šeit arī jāveic cita diagnoze ar laringoskopiju vai noteiktiem Rentgena izmeklējumi. Kvalificētā rehabilitācijas klīnikā muskuļu slimību ārstēšanai svarīgs priekšnoteikums ir holistiska terapeitiska pieeja šai slimībai.
Lielākā daļa muskuļu distrofiju ir labi pētītas klīniski un Detalizēts apraksts izgatavots pagājušā gadsimta beigās. Bet, neskatoties uz gandrīz gadsimtu ilgo muskuļu distrofiju izpētes vēsturi, jautājumi par to patoģenēzi un ārstēšanu joprojām nav atrisināti. Lielas cerības tiek liktas uz molekulāro ģenētiku, ar kuras palīdzību ir noteikta gēnu atrašanās vieta daudzām nosoloģiskām formām.
Tas ietver katedras absolventus psiholoģijas jomā. Skartās personas cieš no fiziskiem trūkumiem un ar to saistītām sekām, jo īpaši — paredzamais dzīves ilgums bieži ir saistīts ar depresīvi noskaņojumi un psihosomatiskās sūdzības. Šeit ļoti noderīgas ir atbalstošas individuālas sarunas vai relaksācijas paņēmienu apguve, piemēram, autogēna apmācība vai progresīva muskuļu relaksācija.
Jautājumi par likumu par māsu, stingra invaliditāte, darba vieta, sazināšanās ar darba devēju profesionālajai rehabilitācijai vai integrācijai uzņēmumā, šeit kā biežas tēmas sociālajā konsultēšanā jāmin. Turklāt bieži vien ir mērķtiecīga konsultēšana konsultācijās izplatīts jautājums pacientiem ar muskuļu traucējumiem. Visbeidzot, ārstēšanas plāns rehabilitācijas laikā Bad Wildungen ietver arī kvalificēta ortopēdijas speciālista koordināciju. Vīzereks ir ortopēdijas nodaļas galvenais ārsts Hombergas klīnikā Bādvildungenā.
Muskuļu distrofijas diagnostika bieži ir ļoti sarežģīta. Klīniskās izpausmes ir ļoti dažādas, un nelielais ģimenes locekļu skaits apgrūtina mantojuma veida noteikšanu.
Raksturīgs motora defekts pacientiem ar muskuļu distrofiju ir “pīles” gaita: pacients staigā, brienot no vienas puses uz otru. Tas galvenokārt ir saistīts ar sēžas muskuļu, galvenokārt vidējo un mazo muskuļu, vājumu, kas fiksē iegurni attiecībā pret augšstilba kaulu. Rezultātā slimība izraisa iegurņa sasvēršanos neatbalstošās kājas virzienā (Trendelenburgas fenomens) un rumpja kompensējošu slīpumu pret pretējā pusē(Dišena fenomens). Ejot, slīpuma puse pastāvīgi mainās. Šīs izmaiņas var pārbaudīt arī Trendelenburgas testā, palūdzot pacientam pacelt vienu kāju, saliekot to taisnā leņķī pie ceļa un gūžas locītavām: paceltās kājas pusē iegurnis nolaižas (un nepaceļas kā parasti) nesošās kājas gluteus medius muskuļa vājuma dēļ.
Klīnikā notiek regulāras apmācības, kā arī dalība attiecīgos kongresos un simpozijos. Šī tikšanās notika jo īpaši. Bieži tiek novērots arī muskuļu enzīmu pieaugums. Slimības parasti attīstās lēni, bet strauji attīstās. Ja jums ir aizdomas, ka jums ir miopātija vai esat saistīts ar zināmu neiromuskulāru slimību, varat ieplānot tikšanās.
Ārstēšana ir atkarīga no konkrēts iemesls, muskuļu iekaisuma slimība var, piemēram, Piemēram, dažādas imūnterapijas. Detalizēts apskats Neiromuskulāro slimību gēnu tabulās var atrast dažādus ģenētiskus muskuļu traucējumus.
Paceļoties no horizontāla stāvokļa, pacients ar izteiktu proksimālo muskuļu vājumu gandrīz nepagriežas uz vēdera, pēc tam, atspiedis rokas pret grīdu, nostājas četrrāpus un pēc tam, atspiedis rokas uz apakšstilbiem, tad uz augšstilbiem. , pamazām iztaisnojas. Šo “iegūšanas pašam” fenomenu sauc par Goversa manevru. Tas bieži ir saistīts ar gluteus maximus muskuļu vājumu.
Zāles, kas var izraisīt vai pastiprināt muskuļu slimības
Mūsu praksē ir pieejamas visas nepieciešamās diagnostikas iespējas. Tas ietver praksi un regulāru apmācību neiromuskulāro slimību jomā. Skartās personas var reģistrēties arī pacientu reģistrā. Pacientu reģistri ir nozīmīgs pirmais solis ceļā uz labāku slimību izpratni un terapeitiskās attīstības pētniecību.
Sirds muskuļa bojājums
Šeit ir apskatīta iespējamā sirds muskuļa iesaistīšanās miopātijās un citās neiromuskulārās slimībās.Dišēna muskuļu distrofija. Dišēna pseidohipertrofiskā muskuļu distrofija rodas biežāk nekā visas citas muskuļu sistēmas slimības (30 uz 100 000 dzīviem jaundzimušajiem). Raksturīga agrīna parādīšanās un ļaundabīga gaita. Klasiskā aina izpaužas kā bērna gaitas izmaiņas 2-5 gadu vecumā, 8-10 gadu vecumā bērni jau staigā ar grūtībām, 14-15 gadu vecumā viņi ir kā likums, pilnībā imobilizēts. Bērniem ir vairāk agrīnā vecumā sākotnējie simptomi izpaužas ar aizkavētu motorisko attīstību: viņi sāk staigāt vēlāk un nevar skriet vai lēkt. Pacienti mirst 2-3 dzīves desmitgadē.
Mēs domājam visas neiromuskulārās slimības. Saskaņā ar Voltona klasifikāciju ir 800 veidlapas. Šis simptoms var attiekties tikai uz dažām muskuļu grupām vai, atkarībā no slimības, var būt visa muskulatūra. Muskuļu novājēšana, samazināšanās muskuļu masa Un muskuļu vājums ir galvenie neiromuskulāro slimību simptomi. Zāles sauc par muskuļu atrofijas mazināšanu. Tomēr tie vēl nenodrošina tiešu diagnozi. Ir vairāki iemesli, kas var izraisīt daļēju muskuļu masas zudumu, kas daļēji ir saistīts ar visu ķermeni.
Viena no pirmajām slimības pazīmēm ir ikru muskuļu sabiezēšana un pakāpeniska to apjoma palielināšanās pseidohipertrofijas dēļ. Augšstilbu un iegurņa jostas muskuļu atrofiju bieži maskē labi attīstīti zemādas taukaudi. Pakāpeniski process iegūst augšupejošu virzienu un izplatās ārpus plecu joslas, muguras muskuļiem un pēc tam uz roku proksimālajām daļām.
Patiesais šādas muskuļu atrofijas vai muskuļu vājuma cēlonis var būt attālās, pilnīgi citās ķermeņa zonās. Cēloņi var būt nervu šūnās muguras smadzenes, kas atbild par kustību, barojošajos nervos, pārnesot nervu uz muskuļu vai pašos muskuļos.
Neiromuskulāro slimību ārstēšana
Ir daudzsološas zāles miozīta, myasthenia gravis un endokrīnās miopātijas ārstēšanai. Pirmās pieejas palēnināšanai ir iespējamas ar laterālā skleroze amiotrofiski. Iedzimtu muskuļu distrofiju un mugurkaula muskuļu atrofiju gadījumā cēloņsakarība vēl nav noteikta. Uz nākotni vērstas īpašas cerības gēnu terapija. Visu neiromuskulāro slimību simptomatoloģija sastāv no konsekventa fizioterapeitiskā atbalsta kombinācijā ar ortopēdiskiem pasākumiem, dažos gadījumos ar elpošanas atbalstu un dažos gadījumos ar sirds transplantācijas iespēju.
IN termināla stadija muskuļu vājums var izplatīties uz sejas, rīkles un elpošanas muskuļiem.
Progresīvā slimības stadijā ir tādi raksturīgie simptomi, kā" pīļu pastaiga”; uzsvērta jostas lordoze, spārnoti plecu lāpstiņas, “vaļīgas plecu jostas” simptoms. Raksturīgas ir agrīnas muskuļu kontraktūras un cīpslu ievilkšanas, īpaši Ahileja cīpslas. Ceļu refleksi pazūd agri, un pēc tam refleksi no augšējo ekstremitāšu.
Ārstēšana ietver arī fizisko ierobežojumu kompensāciju ar pielāgojumiem. Vācijas muskuļu slimību biedrība ir izveidojusi neatkarīgu resursu centru. Tāpat ir iespēja pārbaudīt produktus mājās divos sērijveida dzīvokļos vai pavadīt tikai dažas dienas bezbarjeru dzīvokļos.
Muskuļu darbības traucējumi Neatkarīgi traucējumi Muskuļu relatīvi reti. No otras puses, bieži gadās, ka muskuļus ietekmē citas pamatslimības, īpaši slimības nervu sistēma; Infekcijas slimības piemēram, tīfs un tuberkuloze; Parazitāras slimības, Trīsvienības un cūku tārpi, kā arī hormonālie traucējumi.
Pseidohipertrofija var attīstīties ne tikai ikru muskuļos, bet arī sēžas muskuļos, deltveida muskuļos, vēdera un mēles muskuļos. Ļoti bieži sirds muskulis cieš no kardiomiopātijas veida. Sirds darbības ritma traucējumi, sirds robežu paplašināšanās, toņu kurlums, EKG izmaiņas. Akūta sirds mazspēja ir visvairāk kopīgs iemesls letāli iznākumi Dišēna muskuļu distrofijā. Autopsijā tiek konstatēta sirds muskuļa fibroze un tauku infiltrācija.
Vienkāršs muskuļu zudums bieži notiek konservācijas vai atpūtas rezultātā.Muskuļu kontrakciju var izraisīt saaugumi vai traumas sliktas asinsrites dēļ - Asociāciju nožņaugšanās rezultātā. Muskuļu sāpju cēlonis var būt muskuļu reimatisms, pārslodze, saaukstēšanās, vielmaiņas traucējumi vai atsevišķu muskuļu grupu pārslodze skeleta deformācijas dēļ.
Mioma - labdabīgs audzējs muskuļu audi. Polimiozīts: muskuļu slimība ar līdzīgiem simptomiem kā dermatomiozīts, bet bez ādas izsitumi. Runājot par cēloni, tiek pieņemts, ka traucējumi attiecas uz autoagresiju, kā ciešanām, kas rodas ķermeņa uzbrūkot savām struktūrām.
Bieži tiek novēroti kuņģa-zarnu trakta motorikas traucējumi.
Bieži sastopams simptoms ir intelekta samazināšanās. Interesanti, ka dažās ģimenēs garīgā atpalicība ir izteikta, citās tā ir samērā mērena. Augstāko garīgo funkciju izmaiņas parasti neprogresē un nekorelē ar muskuļu defekta smagumu. To nevar izskaidrot tikai ar slimo bērnu pedagoģisko nolaidību, kuri agri tiek izslēgti no bērnu pulciņiem un neapmeklē bērnudārzs un skola motora defektu dēļ. CT un MRI bieži atklāj smadzeņu atrofiju, kas, iespējams, ir saistīta ar pirmsdzemdību smadzeņu attīstības traucējumiem.
Ir vairāk nekā 200 muskuļu slimību veidu, daži nosaukti to atklājēju vārdā, bet citi pēc slimības. Visizplatītākās ir trīs apakšgrupas. Progresējoša muskuļu distrofija Spināla muskuļu atrofija Neirāla muskuļu atrofija. Klīniskajā praksē šīs slimības ir reti sastopamas un ir absolūti nepieciešamas neirologa rokās.
19. hromosomas ģenētiskais defekts ir bojājums muskuļu šūnas, ko nevar ārstēt līdz šai dienai. Iemesls, iespējams, ir muskuļu šūnu membrānas sistēmas izmaiņas. Mistiskā distrofija var progresēt līdz pilnīgai muskuļu šūnu iznīcināšanai. Pirmkārt, muskuļi tiek ietekmēti sejā, rokās, apakšdelmos, apakšstilbos un pēdās. Slimība var rasties visu vecumu vīriešiem un sievietēm.
Bērniem bieži attīstās adiposogenitālais sindroms un dažreiz arī citas endokrīnās mazspējas pazīmes. Bieži tiek konstatētas izmaiņas skeleta sistēmā: pēdu deformācija, krūtis, mugurkaula, difūzā osteoporoze.
Atšķirīga iezīme Duchenne forma ir augsta pakāpe hiperenzīmi jau ir plkst agrīnās stadijas procesa attīstība. Tādējādi muskuļu audiem specifiskā enzīma - kreatinīna fosfokināzes - līmenis asins serumā var pārsniegt desmitiem un pat simtiem reižu. normāli rādītāji. Straujam (10–100 reižu) kreatinīna fosfokināzes (CPK) palielinājumam neiromuskulārās patoloģijas gadījumā jārosina diskusija galvenokārt par šādām slimībām: Dišēna slimība, Bekera slimība, poliomiozīts un dermatomiozīts, paroksizmāla mioglobulinūrija, distālā miodistrofija. Tikai progresējošās slimības stadijās pakāpeniski samazinās hiperenzimēmijas pakāpe. Ir ziņojumi par palielinātu CK intrauterīnās attīstības stadijā.
Aizkavēta muskuļu relaksācija ir raksturīga pēc muskuļu sasprindzinājums. Sekas ir muskuļu vājums un kustību ierobežojumi kājās, rokās un rokās, kā arī smalko motoriku pasliktināšanās. Daži slimi muskuļi ir īpaši spēcīgi, jo pamatā ir tipisks audu sastāvs muskuļu šķiedras iestrādāts taukos un saistaudos.
To sauc arī par muguras smadzenēm, kas ietekmē muskuļu spazmas. Pastāv līdz 30 dažādas formas mugurkaula muskuļu atrofija. Visizplatītākā forma ir proksimālā mugurkaula muskuļu atrofija, kas aprakstīta šeit. Pēc fizelāžas sākuma tas tiek apzīmēts šādi.
Dišēna muskuļu distrofija tiek pārnesta ar X saistītā recesīvā veidā. Gēns ir lokalizēts X hromosomas īsajā rokā. Gēnu mutāciju biežums ir diezgan augsts (30%), kas izskaidro liels skaits sporādiski gadījumi.
Mutācija (visbiežāk dzēšana) izraisa seksuālu vai gandrīz pilnīgu gēna produkta - distrofijas strukturālā proteīna - neesamību. Fizioloģiskā loma distrofija nav pilnībā noteikta. Tas ir atrodams lielā koncentrācijā sarkolemmas reģionā, acīmredzot spēlējot noteiktu lomu šīs membrānas integritātes saglabāšanā. Distrofijas cēloņu trūkums strukturālās izmaiņas sarkolemmā, kas savukārt izraisa intracelulāro komponentu zudumu un palielinātu kalcija pieplūdumu, kas galu galā izraisa miošķiedru nāvi. Tiek uzskatīts, ka garīgās atpalicības cēlonis ir distrofijas trūkums kortikālo neironu sinaptiskajās zonās.
Cēlonis, iespējams, ir ģenētisks defekts. Nervu šūnas muguras smadzenēs ir inficētas priekšējā raga šūnas. Tiek uzskatīts, ka cēlonis ir nervu nervu sistēma. Tas attiecas tikai uz motoro nervu sistēmu. Nervu sistēmas daļas, kas ir atbildīgas par pieskārienu, sāpēm un temperatūru, paliek neskartas. Funkcija Urīnpūslis un taisnās zarnas nepasliktinās.
Apzīmējums apzīmē arī muskuļu zudumu, ko izraisa nervi. Cēlonis gandrīz vienmēr ir ģenētisks defekts, nervu apvalki kļūst neparasti biezi vai paši nervu apvalki pasliktinās. Ieslēgts nervu šķiedras ietekmē rokas un kājas.
Medicīniskās ģenētiskās konsultācijas vajadzībām ir ļoti svarīgi noteikt heterozigotu pārvadāšanu. Ar Dišēna muskuļu distrofiju heterozigotiem, aptuveni 70% gadījumu subklīniska un dažreiz pat acīmredzamas pazīmes muskuļu patoloģija - neliels ikru muskuļu sabiezējums un pat palielināšanās, ātrs muskuļu nogurums fiziskās aktivitātes laikā, izmaiņas EMG un muskuļu biopsiju patomorfoloģiskajā izmeklēšanā. Visbiežāk heterozigotiem nesējiem uzrāda kreatinīna fosfokināzes aktivitātes palielināšanos.
Nervu vadīšanas ātruma intensitāte palēninās. Sākas apakšējās ekstremitātēs ar muskuļu zudumu un ar to saistītu muskuļu vājumu. Pēc tam simptomi progresē līdz apakšējām ekstremitātēm, vēlāk skarot rokas un apakšdelmus. Jušanas traucējumi ir zemi. Iespējami autonomi traucējumi, piemēram, pārāk daudz vai pārāk maza sviedru veidošanās un bazālās asinsrites traucējumi. Var rasties nelieli krampju simptomi kājās.
Muskuļu vājums rodas muskuļu vājuma dēļ, līdz muskuļi ir pilnībā funkcionāli, kā rezultātā rodas ievērojami, nopietni kustību ierobežojumi kājās, rokās un rokās. Muskuļi ir ļoti svarīga mūsu ķermeņa daļa. Bez muskuļiem ķermenis zaudē spēju kustēties un veikt dažādas aktivitātes. Patiesībā bez cilvēka muskuļu sistēma, jūs, iespējams, nevarēsit izdzīvot. Tas ir saistīts ar faktu, ka lielākā daļa orgānu atrodas gremošanas sistēma sastāv no muskuļiem un pat pati sirds, kas sūknē asinis, arī ir muskulis.
Ja sievietēm ir Dišēna muskuļu distrofijas klīniskā aina, vispirms jāizslēdz anomāliju iespējamība X hromosomā - Šereševska-Tērnera sindroms (TS), Morisa sindroms (XY) vai šo sindromu mozaīcisms.
Dišēna muskuļu distrofija, kas sāk attīstīties pirmsdzemdību periodā, būtībā ir iedzimta miopātija, un to var diagnosticēt drīz pēc dzimšanas, veicot muskuļu biopsiju un nosakot CPK aktivitāti.
Bekera muskuļu distrofija. Līdzās smagajai, ļaundabīgajai ar X saistītās Dišēna muskuļu distrofijas formai ir arī labdabīga forma – Bekera slimība. Pēc klīniskajiem simptomiem tas ir ļoti līdzīgs Dišēna formai, bet, kā likums, sākas vēlāk - 10-15 gadu vecumā, plūst viegli, pacienti ilgstoši paliek darbspējīgi, 20 gadu vecumā. 30 gadus un vēlāk viņi joprojām var staigāt. Auglība nesamazinās, tāpēc slimība dažkārt izsekojama vairākās ģimenes paaudzēs: slims vīrietis ar meitas starpniecību slimību nodod mazdēlam (“vectēva efekts”). Sākotnējie simptomi, tāpat kā Dišēna slimības gadījumā, izpaužas kā vājums iegurņa joslas muskuļos, pēc tam apakšējo ekstremitāšu proksimālajās daļās. Pacientu gaita mainās, viņiem ir grūtības kāpt pa kāpnēm vai piecelties no zema sēdekļa. Raksturīga ir ikru muskuļu pseidohipertrofija. Papēža (Ahileja) cīpslu ievilkšana ir mazāk izteikta nekā Dišēna slimības gadījumā.
Šajā formā nav intelektuālu traucējumu, kardiomiopātijas nav vai tā ir nedaudz izteikta.
Tāpat kā ar citām X saistītām miodistrofijām, Bekera formā CPK aktivitāte ievērojami palielinās, lai gan mazākā mērā nekā Dišēna slimības gadījumā, nepārsniedzot 5000 vienības. Bekera slimības gēns, tāpat kā Dišēna slimība, ir lokalizēts X hromosomas īsajā rokā; iespējams, ka abi loki ir cieši saistīti vai alēli. Atšķirībā no Dišēna slimības, kurā distrofijas praktiski nav, Bekera slimībā tiek sintezēta patoloģiska distrofija. Atšķirības tiek konstatētas arī muskuļu biopsijā. Bekera muskuļu distrofijas gadījumā muskuļu šķiedras parasti ir nenoapaļotas, Dišēna muskuļu distrofijai raksturīgās hialīna šķiedras tiek novērotas ārkārtīgi reti.
Landouzy-Dejerine miodistrofija (facioscapulohumeral miodistrofija). Slimību pārnēsā autosomāli dominējošā veidā ar augstu penetranci, bet nedaudz mainīgu ekspresivitāti. Tas ir daudz retāk nekā Dišēna muskuļu distrofija (0,4 uz 100 tūkstošiem iedzīvotāju). Tiek uzskatīts, ka šīs slimības gēns ir lokalizēts 4. hromosomā. Sievietes slimo biežāk nekā vīrieši (3:1), Fiziska pārslodze, intensīva sportošana, kā arī neracionāla fizioterapija var veicināt smagāku slimības gaitu.
Landouzy-Dejerine muskuļu distrofija ir salīdzinoši labvēlīga pašreizējā muskuļu patoloģijas forma. Tas sākas apmēram 20 gadu vecumā, dažreiz vēlāk. Savukārt ģimenes saslimšanas gadījumos, kad iespējams sekot līdzi gados jaunākiem ģimenes locekļiem, kādu muskuļu vājumu, piemēram, sejas muskuļus, iespējams konstatēt jau agrāk.
Muskuļu vājums un atrofija vispirms parādās sejas muskuļos vai plecu josta. Pakāpeniski šie traucējumi izplatās uz proksimālo roku muskuļiem un pēc tam uz apakšējās ekstremitātes. Vairumā gadījumu vispirms tiek ietekmēti kāju priekšējās virsmas muskuļi (attīstoties pēdas kritumam), pēc tam proksimālo kāju muskuļi. Slimības augstumā tiek smagi ietekmēti acs un mutes apļveida muskuļi, lielā krūšu kurvja, serratus anterior un apakšējā trapecveida muskulatūra, latissimus dorsi, biceps, triceps brachii muskuļi. Raksturīgs izskats pacienti: tipiska seja miopātija ar "šķērsvirziena smaidu" ("Gioconda smile"), izvirzījums augšlūpa(“tapīra lūpas”), izteikti spārnveidīgi plecu lāpstiņas, savdabīga krūškurvja deformācija ar tās saplacināšanu anteroposterior virzienā un griešanos uz plecu locītavu iekšpusi. Bojājumam bieži ir asimetrija pat vienā muskulī (piemēram, orbicularis oris muskulī). Var novērot ikru, deltveida muskuļu un dažreiz arī sejas muskuļu pseidohipertrofiju. Kontrakcijas un ievilkšanas ir mēreni izteiktas. Cīpslu refleksi ilgu laiku saglabājas, bet dažkārt samazinās jau agrīnā stadijā.
Sirds muskuļa bojājumu pazīmes tiek atklātas reti. Seruma enzīmu aktivitāte ir nedaudz palielināta un var būt normāla. Intelekts necieš. Dzīves ilgums vairumā gadījumu nesamazinās. Interesanti, ka EMG Landouzy-Dejerine miodistrofijas gadījumā bieži vien nav pilnībā raksturīgs bojājuma muskuļu līmenim. Dažiem pacientiem (vienas ģimenes locekļiem) var rasties biopotenciālu amplitūdas samazināšanās, traucējumu veida līkne; citiem, gluži pretēji, biežuma un hipersinhronās aktivitātes samazināšanās, dažreiz ar tipisku palisādes ritmu. Jāapzinās mugurkaula variants, kas atdarina Landouzy-Dejerine slimību.
Erb-Roth muskuļu distrofija (ekstremitāšu jostas muskuļu distrofija). Tas tiek pārraidīts autosomāli recesīvā veidā, un abi dzimumi tiek ietekmēti vienādi. Slimības sākums vairumā gadījumu datējams ar 2. dzīves desmitgades vidu (14-16 gadi), taču tā tiek raksturota kā agrīna, pseido-dišēna forma, kad pirmie simptomi parādās līdz 10 gadu vecumam. un slimība ir smaga, un vēlīna versija ar sākumu pēc 30 gadiem.
Slimības gaita var būt ātra vai lēnāka, vidēji pilnīga invaliditāte iestājas 15-20 gadus no pirmo simptomu parādīšanās. Miodistrofija sākas vai nu ar iegurņa jostas un proksimālo kāju muskuļu bojājumiem (Leiden-Moebius forma), vai no plecu jostas (Erb forma). Dažos gadījumos plecu un iegurņa jostas tiek ietekmētas vienlaikus. Diezgan ievērojami cieš muguras un vēdera muskuļi. Pacientiem ir raksturīga “pīļu” gaita, grūtības piecelties no guļus vai sēdus stāvokļa, uzsvēra jostas lordoze. Vairumā gadījumu sejas muskuļi netiek ietekmēti. Kontrakcijas un pseidohipertrofijas šai formai ir retāk sastopamas. Var rasties termināla atrofija un cīpslu ievilkšana. Intelekts parasti tiek saglabāts. Sirds muskuļi lielākoties netiek ietekmēti. Seruma enzīmu līmenis parasti ir paaugstināts, bet ne tik dramatiski kā ar X saistītas muskuļu distrofijas gadījumā. Ir norādes, ka vīriešiem vīriešiem ir augstāks KFK līmenis nekā sievietēm. Pastāv būtiska atšķirība mutanta gēna ekspresijā starp dažādiem ģimenes locekļiem - kopā ar smagiem klīniskā aina Var būt salīdzinoši viegli un pat neskaidri klīniskie simptomi. Nāve parasti notiek no plaušu komplikācijām.
Tā kā ekstremitāšu jostas miodistrofijas klīniku īpaši viegli atdarina dažāda rakstura neiromuskulārās slimības, tas ir nepieciešams, īpaši sporādiskos gadījumos un ar vēls sākums slimības, veiciet uzmanīgi klīniskā pārbaude izslēgt mugurkaula amiotrofiju, polimiozītu, vielmaiņas, endokrīnās, toksiskās, zāļu izraisītās, karcinomatozās miopātijas. Agrāk šai muskuļu distrofijas formai ir bijusi skaidra pārmērīga diagnoze.
Muskuļu distrofiju ārstēšana. Muskuļu distrofiju terapijas iespējas ir ļoti ierobežotas. Etioloģiskās un patoģenētiskā ārstēšana praktiski neeksistē. Simptomātiskā ārstēšana galvenokārt ir vērsta uz kontraktūru attīstības novēršanu, esošā muskuļu spēka saglabāšanu un, iespējams, nedaudz samazinātu atrofijas attīstības ātrumu. Galvenais uzdevums ir maksimāli palielināt periodu, kurā pacients spēj patstāvīgi pārvietoties, jo guļus stāvoklī ātri palielinās kontraktūras, skolioze un elpošanas traucējumi. Medicīnas komplekss jāiekļauj ārstnieciskā vingrošana, masāža, ortopēdiskie pasākumi un zāļu terapija.
Ārstnieciskā vingrošana sastāv no pasīvām un aktīvām kustībām, ko veic visās locītavās dažādās pozīcijās: stāvus, sēdus, guļus, ar dažādām ekstremitāšu pozīcijām. Aktīvās kustības Vēlams veikt izometriskā režīmā. Vingrošanas nodarbības jāveic regulāri vairākas reizes dienā. Tajā pašā laikā jāievēro piesardzība pret pārmērīgu slodzi, īpaši, ja to pavada muskuļu pārstiepums. Svarīgi ir elpošanas vingrinājumi (īpaši pēc pacienta imobilizācijas).
Ortopēdiskie pasākumi Konservatīvās (īpašas šinas) un ķirurģiskās (ahilotomija, gastrocnemius muskuļa šķērsgriezums), kuru mērķis ir koriģēt kontraktūras un jaunus patoloģiskos ekstremitāšu izlīdzinājumus, ir arī mērķis saglabāt spēju patstāvīgi pārvietoties. Katrā gadījumā ir nepieciešams individuāli izsvērt sagaidāmos ieguvumus un iespējamo kaitējumu no operācijas. Jāņem vērā, ka bieži (īpaši ar smagu hiperlordozi un četrgalvu augšstilba muskuļa vājumu) pēdu ekvinovarusam stāvoklim ir kompensējoša vērtība un pēc, piemēram, ahilotomijas pacients var būt pilnībā imobilizēts. Kontraktūru attīstīšanai ieteicams maigi izstiept muskuļus līdz 20-30 reizēm dienā, pēc tam miega laikā uzlikt šinu.
Narkotiku terapija ietver vielmaiņas zāļu izrakstīšanu, kuru mērķis ir atjaunot enerģijas un olbaltumvielu deficītu, taču to efektivitāte ir ļoti apšaubāma. Tiek izmantoti kalcija antagonisti (Dišena slimības konstatētā defekta dēļ šūnu membrānas, kas izraisa palielinātu kalcija iekļūšanu šūnā), imūnmodulatori, fosforu saturoši savienojumi (ATP, fosfadēns), E vitamīns (100 mg iekšķīgi 3 reizes dienā). Ir pierādīts, ka Dišēna slimības gadījumā prednizolona lietošana (0,75 mg/kg dienā) var ievērojami palielināt muskuļu spēku, taču šis efekts ilgst ne vairāk kā gadu un kopumā neietekmē slimības iznākumu. Sakarā ar nopietnu blakus efekti, rodas meli ilgstoša lietošana narkotiku, to lietošana nav piemērota. Efektu aplēses anaboliskie steroīdi ir pretrunīgi, un to mērķis bieži ir saistīts ar nepamatotu risku. Novērtējot noteiktu zāļu iedarbību Dišena slimības gadījumā, jāņem vērā, ka ar vidēji smagu slimības smagumu 3-6 gadus veciem pacientiem var būt relatīva stāvokļa stabilizēšanās, kas saistīta ar ar vecumu saistītu muskuļu attīstību. sistēma, motorisko prasmju apguve, kas zināmā mērā var īslaicīgi kompensēt nepārtraukti notiekošo deģeneratīvo procesu.
Īpaši svarīga ir pacienta uztura korekcija, ieteicama diēta ar augstu olbaltumvielu, zemu tauku un kaloriju daudzumu ar optimālu vitamīnu un mikroelementu saturu. Svarīga loma ir pacienta psiholoģiskajam atbalstam, tālākizglītībai un atbilstošai profesionālai vadībai.
44. lapa no 44
Skeleta muskuļi ir iesaistīti patoloģisks process par dažādām deģeneratīvām, vielmaiņas un iekaisuma slimības. Vairumā gadījumu tas izraisa muskuļu šķiedru deģenerāciju un hroniskās formās to nomaiņu saistaudi un tauki. Proksimālās muskuļu grupas ir vairāk bojātas nekā distālās, tāpat kā apakšējās ekstremitātes attiecībā pret augšējo. Slimam bērnam ir tā sauktā pīlei līdzīga gaita, un viņš sēdus stāvoklī nespēj skriet, kāpt pa kāpnēm vai piecelties kājās. Viņa cīpslu refleksi ir nomākti, to izzušanas pakāpe ir proporcionāla muskuļu spēka pavājināšanās pakāpei. Jutība nav traucēta.
Diagnostiski vērtīgas laboratorijas metodes ietver enzīmu, īpaši kreatīnfosfokināzes, aktivitātes noteikšanu serumā. Šis enzīms, kas katalizē reakciju: fosfokreatīns + ADP-kreatīns + ATP, atrodas galvenokārt smadzeņu šūnās un muskuļu audos. Dažās difūzās muskuļu slimībās, īpaši muskuļu distrofijas gadījumā, tās pārmērīgais daudzums iekļūst starpšūnu telpā un asinīs. Pacientiem parasti ir paaugstināta seruma laktātdehidrogenāzes un glutamīnoksaloetiķskābes transamināzes aktivitāte, bet to plaša izplatība citos audos, tostarp aknās, samazina testa specifiku. Parasti diagnozes precizēšanai ir nepieciešama muskuļu audu biopsija.
Muskuļu iekaisuma slimības. Muskuļu audu iekaisums pavada dažas infekcijas, īpaši trihinelozi, toksoplazmozi un Koksaki vīrusa izraisītas infekcijas. Tas bieži ir kolagēna slimību sastāvdaļa, tostarp dermatomiozīts, sarkanā vilkēde, mezglains periarterīts un reimatoīdais artrīts.
Polimiozīts. Izkliedētu izolētu nezināmas etioloģijas muskuļu iekaisumu sauc par polimiozītu. To raksturo strauja progresējoša gaita, vājums un sāpes proksimālajās muskuļu grupās. Bieži procesā tiek iesaistīti kakla muskuļi, kas apgrūtina bērna galvas pacelšanu un noturēšanu šajā stāvoklī. Muskuļu iekaisuma laboratoriskās pazīmes ir ESR un balto asins šūnu skaita palielināšanās. Tomēr to trūkums neizslēdz polimiozītu. Seruma enzīmu līmenis parasti ir paaugstināts. Muskuļu biopsijā nosaka šķiedru deģenerāciju un daļēju atjaunošanos un to infiltrāciju ar limfoīdām šūnām. Ir grūti atšķirt polimiozītu no muskuļu distrofijas un dermatomiozīta. Tas var būt netipisks dermatomiozīta veids, lai gan šo divu stāvokļu histoloģiskais attēls ir nedaudz atšķirīgs: dermatomiozītu raksturo vaskulīts, kura polimiozīta gadījumā parasti nav. Pēdējā prognoze ir nedaudz labvēlīgāka. Ārstēšana ar kortikosteroīdiem ir saistīta ar efektu, bet, tos pārtraucot, var rasties recidīvs.
Progresējošs ossificans miozīts. Šīs retās saistaudu un muskuļu slimības etioloģija nav zināma. Tiek ziņots, ka tas ietekmē brāļus un māsas, tostarp dvīņus, un tiek nodots asinsradiniekiem tiešā līnijā. Tiek pieņemts, ka slimība tiek mantota autosomāli dominējošā veidā. Zēni slimo 2-3 reizes biežāk nekā meitenes.
Patoloģiskās pazīmes ir atkarīgas no slimības stadijas. Agrīnās stadijās muskuļos un cīpslās konstatē lokālu pietūkumu un iekaisuma šūnu infiltrātus. Vēlāk iekaisuma zonas tiek aizstātas ar granulācijas audiem, un galu galā bojājumos veidojas skrimšļa un kaulu audu zonas.
Gandrīz 75% slimo bērnu tiek diagnosticēti dzimšanas defekti attīstība, visbiežāk roku pirkstu nepietiekama attīstība un pirmo kāju pirkstu falangu ankiloze un pirmo pirkstu nepietiekama attīstība, polidaktilija, pirkstu izliekums, sindaktilija (kājas), deformācija ausis, kurlums, zobu trūkums. Tādi paši iedzimtie defekti var būt arī pacienta radiniekiem, kuriem nav attīstījusies progresējoša saistaudu un muskuļu slimība. Vecums, kurā var sākties miozīts ossificans, atšķiras no dzimšanas līdz vecākai bērnībai. Parasti tam ir trīs stadijas: 1) nelielu lokālu traumu vietās parādās ierobežoti, nereti silti un pēc taustes mīkstai mīklai līdzīgi mīksto audu pietūkumi; 2) pēc dažām dienām izzūd iekaisuma simptomi, un bojājums sacietē; 3) notiek skartās vietas pārkaulošanās. Periodiski parādās jauni bojājumi, galvenokārt kakla un muguras zonās. Primārais simptoms Torticollis var attīstīties, ja process ir attīstījies sternocleidomastoid muskulī. Pārkaulošanās galu galā izplatās uz daudzām cīpslām un saitēm. Rodas mugurkaula un roku un kāju locītavu ankiloze (21.-5. att.). Iekaisums var izplatīties uz temporomandibulārajām locītavām, apgrūtinot košļājamās kustības. Kaulu spuras var izspiesties cauri ādai. Pusaudža gados slimība bieži izraisa pilnīgu nekustīgumu un nāvi elpošanas mazspēja un elpošanas apstāšanās, lai gan ir ziņojumi par izdzīvošanu. Ar miozītu ossificans pastāv augsts osteogēnas sarkomas attīstības risks.
Rīsi. 21-5. Bērns ar progresējošu ossificans miozītu (tipiska poza ar kakla un muguras muskuļu stīvumu).
Dažreiz patoloģiskais process aprobežojas ar iepriekšēju mīksto audu bojājumu vietu (miositis ossificans circumscripta). Plaša muskuļu audu pārkaļķošanās var rasties arī hroniska polimiozīta un dermatomiozīta gadījumā.
rezultātus laboratorijas metodes pētījumiem nav diagnostiskas vērtības.
Kalcija, fosfora, sārmainās fosfatāzes līmenis serumā, kā arī kreatīnfosfokināzes un citu enzīmu aktivitāte paliek normālā līmenī. Kauls bojājuma avotā pēc struktūras neatšķiras no normas.
Esošās metodesārstēšana ir neapmierinoša. Dažos gadījumos, lietojot AKTH un citus kortikosteroīdus, tika novērota slimības progresēšanas palēnināšanās. Viņu loma ārstēšanas gala iznākumā ir apšaubāma.
Endokrīnās un vielmaiņas miopātijas. Miopātija hipertireozes gadījumā ir diezgan reta komplikācija. To raksturo ptoze, divpusēja sejas muskuļu un proksimālo ekstremitāšu muskuļu parēze. Šajā gadījumā dažus hipertireozes simptomus var maskēt muskuļu vājums, bet tahikardija, pastiprināta svīšana un pastiprināta vairogdziedzeris. Cīpslu refleksi atšķirībā no daudzām citām miopātijas formām paliek normāli. Pēc hipertireozes korekcijas muskuļu vājums pakāpeniski izzūd.
Miopātija hipotireozes gadījumā. Hipotireoze zīdaiņiem var būt saistīta ar muskuļu vājumu un hipotensiju. Vecākiem bērniem ar miksedēmu muskuļu kontrakcijas un relaksācijas palēninās, un dažos gadījumos tiek novērota muskuļu hipertrofija (Debreu-Semelen sindroms). Tādu pazīmju kā vājums un muskuļu hipertrofija kombinācija liecina par muskuļu distrofiju.
Miopātija ārstēšanas laikā ar kortikosteroīdiem. Tas var sarežģīt Itsenko-Kušinga slimību, bet biežāk attīstās, ārstējot ar lielām sintētisko steroīdu devām. Īpaši vājums ir jūtams iegurņa jostas muskuļos, kas izpaužas kā bristošā (pīlei līdzīgā) gaita, grūtības kāpt pa kāpnēm un mēģināt piecelties no sēdus stāvokļa. Nav ceļa refleksu. Var rasties muskuļu retināšana. Miopātiskās izmaiņas muskuļu audos parasti ir nenozīmīgas pat ar smagu vājumu. Muskuļu spēks pēc kortikosteroīdu lietošanas pārtraukšanas atveseļošanās notiek lēni (vairāku mēnešu laikā).
Miopātija hiperparatireozes gadījumā. Hiperparatireoze var būt saistīta ar vājumu un hiporefleksiju, ko izraisa hiperkaliēmija. Parasti tie ātri izzūd pēc paratireoidektomijas.
Karnitīna deficīts (lipīdu miopātija) pavada liela daudzuma lipīdu uzkrāšanos muskuļos un, kā rezultātā, traucēta to energoapgāde. Karnitīns ir būtiska sistēmas sastāvdaļa, kas nodrošina taukskābju pārnešanu no gara ķēde no citozola uz mitohondrijiem, kur tie tiek oksidēti. Muskuļu vājums attīstās divos karnitīna deficīta veidos.
Karnitīna deficītu muskuļos klīniski raksturo to proksimālo grupu progresējošs vājums, biežāk skolēniem un pusaudžiem. Dažreiz vājums ir periodisks un apvienots ar mioglobinūriju. Smagos gadījumos var rasties elpošanas muskuļu paralīze. Seruma enzīmu (kreatīnkināzes un aldolāzes) līmenis palielinās. Elektromiogramma atklāj nespecifiskas izmaiņas, kas raksturīgas miopātijai. Muskuļu biopsijā var redzēt lielu skaitu tauku pilienu. Karnitīna līmenis serumā nemainās, bet muskuļu karnitīna līmenis samazinās. Patoloģijas atpazīšana ir būtiska, jo to var izārstēt. To bieži sajauc ar muskuļu distrofiju. Ietekme var rasties pēc 100 mg/(kg/dienā) karnitīna perorālas lietošanas. Dažos gadījumos ārstēšana ar kortikosteroīdiem ir efektīva.
- Sistēmisks karnitīna deficīts izpaužas kā progresējoša miopātija, tostarp kardiomiopātija, un aknu darbības traucējumi, kam pievienota Reja sindroma veida aknu encefalopātijas klīniskā aina. Karnitīna deficīts no pēdējā atšķiras ar tā atkārtotu gaitu un smagu muskuļu vājumu, kas saglabājas starp encefalopātijas saasināšanās periodiem. Seruma kreatīna fosfokināzes līmenis ir ievērojami paaugstināts un karnitīna līmenis pazeminās gan serumā, gan muskuļos. Izmaiņas biopsijas paraugā ir līdzīgas izmaiņām ar karnitīna deficītu muskuļu audos. Līdzīgas klīniskas un morfoloģiskas izmaiņas, tostarp karnitīna deficītu, var konstatēt organisko skābju metabolisma traucējumos, piemēram, metilmalonskābes un glutārā acidūrijā (sekundārais karnitīna deficīts).
Rīsi. 21-6. Bērns ar iedzimtu kreisā krūšu muskuļa trūkumu. 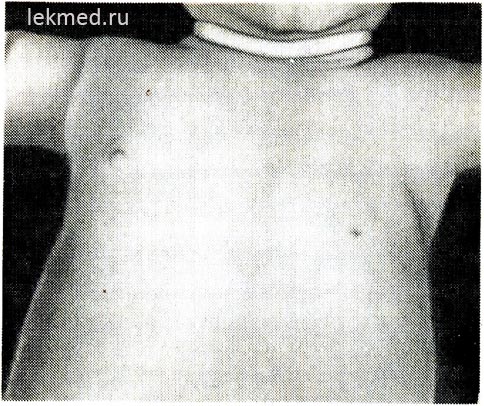
Ievērības cienīgi ir priekšējās paduses krokas un zemā nipelis neesamība.
Ārstēšana sastāv no tā, ka pacients ievēro diētu, kas bagāts ar ogļhidrātiem un ar zemu tauku saturu, un lieto karnitīnu dienas devā 100 mg/kg.
Iedzimti muskuļu defekti. Iedzimta muskuļu trūkums. Muskuļu nepietiekama attīstība var būt diezgan izplatīta parādība un izraisīt pilnīgu locītavu kustību bloķēšanu vai iedzimtu artrogripozi. Kā iedzimts defekts visbiežāk trūkst viena muskuļa. Diezgan izplatīta anomālija ir lielā krūšu muskuļa krūšu daļas trūkums (21.-6. att.), dažos gadījumos šis defekts tiek kombinēts ar sindaktiliju skartajā pusē (Polijas sindroms). Krūšu muskuļa trūkums bieži vien pavada muskuļu distrofiju. Iedzimts vēdera muskuļu trūkums bieži ir saistīts ar urīnceļu attīstības defektiem.
Rīsi. 21-7. Kakla deformācija un sejas asimetrija zēnam ar iedzimtu torticollis, neārstēts kopš 12 gadu vecuma. 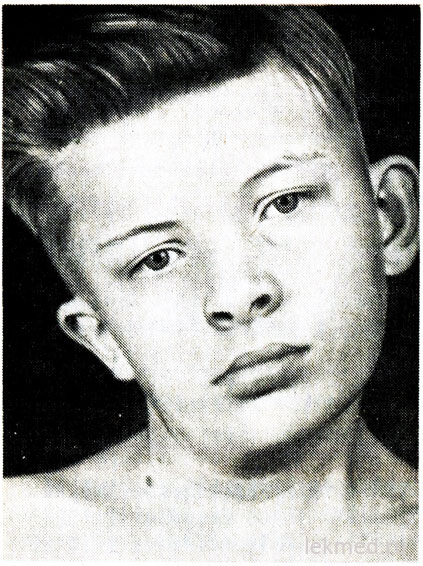
Iedzimtu tortikollisu izraisa sternocleidomastoid muskuļa vienpusējs saīsinājums vai kontraktūra. Pacienta galva ir noliekta pret kontraktūru, un zods ir vērsts uz leju pretējā virzienā (21.-7. att.). Mēģinot koriģēt galvas stāvokli, jūtama ievērojama muskuļu pretestība. Skartajā muskulī ir jūtamas sablīvēšanās vietas. Defekta cēlonis nav skaidrs, ilgu laiku tika uzskatīts, ka tas radās dzimšanas trauma. Tomēr torticollis rodas bērniem, kas dzimuši operācijas rezultātā ķeizargrieziens; tas liecina, ka dažos gadījumos defekta cēlonis ir saistīts ar pirmsdzemdību periodu. Torticollis ir jānošķir no patoloģiska galvas slīpuma kakla skriemeļu deformācijas dēļ, piemēram, ar Klippel-Weil anomāliju, un no kakla skriemeļu lūzumiem vai izmežģījumiem. Tie tiek izslēgti, izmantojot radiogrāfisko izmeklēšanu. Vecākiem bērniem var rasties galvas slīpums ar šķielēšanu, distoniju, aizmugurējā galvaskausa dobuma audzējiem un kakla mugurkauls muguras smadzenes, miozīts ossificans, kakla limfadenīts vai diafragmas trūce. Vairumā gadījumu iedzimtu torticollis var koriģēt ar ārstnieciskā vingrošana. Tomēr, kad hroniska forma torticollis izraisa asimetrisku sejas un galvas attīstību (sk. 21-7. attēlu), kam kosmētisku iemeslu dēļ var būt nepieciešama muskuļu sadalīšana.
Iedzimtas miopātijas. Šajā grupā ietilpst vairākas retas iedzimtu slimību formas, kurās muskuļu vājums un hipotensija parādās jau zīdaiņa vecumā (sk. 22-1. tabulu). Viņu precīza diagnoze ir liela nozīme no prognozes viedokļa. Kopumā tas ir labvēlīgs normālai funkcionēšanai un paredzamajam dzīves ilgumam, atšķirībā no Verdniga-Hofmaņa slimības vai iedzimtas muskuļu distrofijas. Iedzimtu miopātiju noteikšanu parasti atvieglo muskuļu biopsija.
- Centrālās serdes slimība. Muskuļu šķiedru centrālā daļa ir krāsota neparasti, bet vienmērīgi. Elektronu mikroskopiskā izmeklēšana atklāj mitohondriju skaita samazināšanos un sarkoplazmas retikuluma izsīkumu šķiedru centrālajā daļā.
Nemalīna miopātija. Termins "nemalīns" ir izskaidrojams ar to, ka muskuļu šķiedrās ir noteiktas pavedieniem līdzīgas struktūras.
Rīsi. 21-8. Mēles miotoniskā kontrakcija (a) ar asu sitienu ar perkusijas āmuru labajā pusē un plakstiņiem (b) bērnam ar hiperkalēmisku ģimenes periodiskas paralīzes formu. 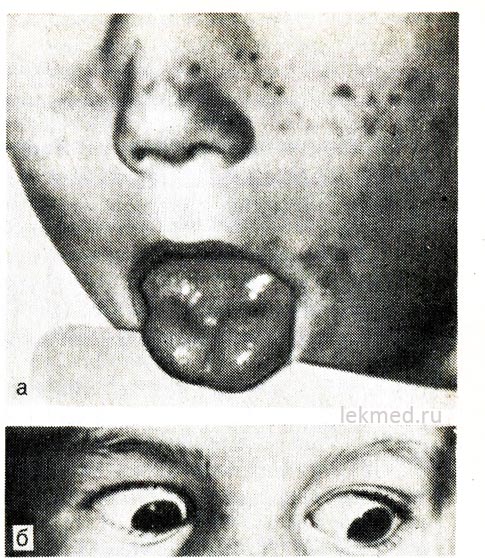
Skatoties uz leju, plakstiņš paliek savilkts.
Elektronu mikroskopiskie dati liecina, ka tas ir miofibrilu Z joslu izmaiņu rezultāts.
Mitohondriju miopātijas. Ir ziņots par dažām miopātiju formām, kurās vissvarīgākās izmaiņas notiek muskuļu šķiedru mitohondrijās. To skaits un izmērs var ievērojami palielināties. Muskuļu vājumu un hipotensiju var konstatēt jau zīdaiņa vecumā, bet dažkārt tie jūtami progresē tikai skolas vecumā. Šīs grupas miopātijas bieži pavada kardiomiopātija, encefalopātija un laktacidēmija.
Miotonija. Šis stāvoklis liecina par dažādām muskuļu slimībām, piemēram, distrofisku miotoniju, ģimenes paroksizmālas paralīzes hiperkalēmisko formu un glikogēna uzkrāšanās slimībām. Miotonija tiek definēta kā ievērojama muskuļu relaksācijas aizkavēšanās pēc brīvprātīgām vai piespiedu kontrakcijām. Klīniski tas izpaužas nespējā atspiest dūri vai redzamā ilgstošā muskuļu kontrakcijā pēc to stimulēšanas, kas izteikta asā kairinājuma veidā (21.-8. att.). To var novērot, ja ar perkusijas āmuru sitat virspusēju muskuļu grupu, piemēram, mēles muskuļus vai plaukstas virsmu pirmā pirksta izciļņa zonā. Miotoniju apstiprina elektromiogrāfijas dati. Šajā gadījumā raksturīga spontāna muskuļu aktivitāte ir pamanāma pēc relaksācijas vai brīvprātīgas kontrakcijas (miotoniskas izdalījumi).
Iedzimta miotonija (Tomsena slimība). Vienīgā šīs slimības pazīme, kas pārmantota pēc dominējošā tipa, ir miotonija. Tas var izpausties zīdaiņa vecumā kā rīšanas kustību palēnināšanās un tai sekojoša vemšana.
nespēja normāli atslābināt rīkles muskuļus. Vecākajos gados bērnība miotonija izpaužas kā pacienta nespēja atvilkt dūrē savilktus pirkstus. Pirmajā mēģinājumā veikt kādu kustību bērna muskuļi kļūst grūti. Daudzas reizes atkārtojot vienu un to pašu kustību, viņi nedaudz atpūšas. Piemēram, slimam bērnam ir lielas grūtības sākt staigāšanu. Viņš parasti pirmos soļus sper ļoti vilcinoši un lēni. Pēc dažām sekundēm gaita kļūst normāla vai gandrīz normāla. Miotonijas simptomus pastiprina nelabvēlīgs pacienta emocionālais stāvoklis un ķermeņa atdzišana. Muskuļu spēks paliek normāls, muskuļi ir pietiekami attīstīti un bieži vien manāmi palielināti, kas rada maldīgu priekšstatu par pacienta sportisko uzbūvi.
Diagnoze pamatojas uz klīniskajiem datiem un elektromiogrāfijas datiem. Seruma enzīmu aktivitāte ir normas robežās. Vienīgā histoloģiskā pazīme ir muskuļu šķiedru hipertrofija.
Slimība atšķiras no distrofiskās miotonijas ar to, ka nav muskuļu vājuma un atrofijas un distrofiskas izmaiņas muskuļu audu biopsijā. Ārstēšana ar novokaīnu vai hinidīna sulfātu ir saistīta ar efektu un ir indicēta funkcionāliem traucējumiem. Parasti slimības gaita ir labdabīga, un līdz ar vecumu pacienta stāvoklis var uzlaboties.
Paroksizmāla paralīze.Šai slimību grupai raksturīgs periodisks muskuļu vājums ar pilnīgu vai gandrīz pilnīgu muskuļu spēka atjaunošanos periodā starp uzbrukumiem. Tas ietver arī muskuļu fosforilāzes deficītu (McArdle slimību).
Hiperkalēmiska paroksizmāla paralīze. Iedzimta epizodiska adinamija jeb paramiotonija tiek pārnesta dominējošā veidā un ir īpaši smaga vīriešiem. Tas parasti sākas agrā bērnībā (dažreiz zīdaiņa vecumā). Uzbrukumi rodas atpūtas periodā pēc lielas muskuļu slodzes. Vājums attīstās ātri un var ilgt vairākas stundas. Tas ir īpaši jūtams kājās; elpošanas funkcija parasti nav traucēta. Bieži vien adinamiju pavada miotonija, kas saglabājas starp uzbrukumiem, kas visspilgtāk izpaužas kā plakstiņu kustības aizkavēšanās, skatoties uz leju (sk. 21.-8. att., b).
Kālija līmenis serumā bieži tiek paaugstināts uzbrukuma laikā, taču, lai to ticami noteiktu, var būt nepieciešama atkārtota pārbaude vairāku uzbrukumu laikā. Uzbrukumu var mākslīgi provocēt, izmantojot kālija slodzi (2-3 g iekšķīgi), taču to drīkst veikt tikai EKG uzraudzībā. Atkārtoti uzbrukumi tiek apturēti ar diakarbu. Smagas slimības formas raksturo hroniska, viegla vājuma un distrofisku izmaiņu attīstība muskuļos.
Hipokalēmiska paroksizmāla paralīze. Ģimenes paroksizmāla paralīze, kas arī dominē dominējošā veidā, ir īpaši smaga zēniem. Atšķirībā no hiperkaliēmiskās formas, pirmais uzbrukums parādās vēlā bērnībā vai agrīnā pusaudža vecumā. Iemesls ir lielu, ogļhidrātiem bagātu maltīšu patēriņš vai atpūta pēc tam fiziskā aktivitāte. Parasti uzbrukums sākas plkst Nākamais rīts pēc smagas fiziskās slodzes un lielām vakariņām. To raksturo muskuļu vājums un arefleksija. Var būt traucēta elpošanas funkcija. Var rasties aritmija, tostarp ventrikulāras ekstrasistoles un tahikardija. Lēkmes var ilgt vairāk nekā 24 stundas.Paralītiskajā fāzē kālija līmenis serumā parasti samazinās (2-3 mmol/l). Pamatdefekts nav zināms. Pacientiem ar atkārtotiem smagiem uzbrukumiem attīstās hronisks muskuļu vājums un patoloģiskas izmaiņas muskuļos. Ārstēšana uzbrukumu laikā sastāv no kālija hlorīda lietošanas; tā sākotnējā deva ir 2-3 g.Diacarb palīdz samazināt lēkmju biežumu.
Paroksizmāla mioglobinūrija (idiopātiska mioglobinūrija). Idiopātiskā mioglobinūrija ir neviendabīga slimību grupa, kurā paralīzes lēkmes ar mioglobinūriju rodas spontāni vai pēc intensīvas fiziskas slodzes. Slimība ir iedzimta dominējošā veidā, saistīta ar X hromosomu. Uzbrukuma laikā muskuļi, visbiežāk ikru un augšstilbu muskuļi, kļūst sāpīgi un pietūkuši. Urīns kļūst tumši sarkans vai Brūna krāsa. Mioglobinūrija var izraisīt nieru kanāliņu nekrozi, kas izraisa nāvi nieru mazspēja.
Diagnozi apstiprina mioglobulīna noteikšana urīnā. Pozitīvs benzidīna tests, ja urīnā nav sarkano asins šūnu, apstiprina mioglobīna klātbūtni tajā, īpaši, ja hemoglobīns serumā nav noteikts. Hemoglobīnu nosaka, izmantojot spektrofotometriju. Paroksizmālā mioglobinūrija ir jānošķir no Makārdla slimības, karnitīna palmitiltransferāzes deficīta un mioglobinūrijas pēc neparastas intensīvas fiziskās aktivitātes vai muskuļu traumas. vesels cilvēks. Mioglobinūrija pēc smagas muskuļu slodzes rodas pseidohipertrofiskas muskuļu distrofijas (Dišena slimības) gadījumā.
Ārstēšana sastāv no gultas režīma; ja nepieciešams, veikt mākslīgā ventilācija plaušas. Lai novērstu nieru mazspēju, ir nepieciešams izrakstīt pacientam daudz šķidruma.
Karnitīna palmitiltransferāzes deficīts. Ja šī enzīma trūkst, tiek traucēta garo ķēžu taukskābju pārnešana uz mitohondriju segmentiem, kuros notiek oksidēšanās un ketonu veidošanās. II tipa izoenzīma deficīts tiek mantots recesīvā veidā. Tā trūkuma dēļ tiek traucēta ketoģenēze audos, tostarp muskuļos un aknās. Pirmās slimības pazīmes biežāk parādās skolas vecuma bērniem un pusaudžiem. Tās sastāv no atkārtotām muskuļu sāpju, vājuma un drudža epizodēm pēc fiziskas slodzes vai badošanās. Mioglobinūrija, ko pavada uzbrukumi, var izraisīt nieru mazspēju. Badošanās izraisa hipoglikēmiju. Starp uzbrukumiem bērni šķiet veseli. Slimība ir jānošķir no citiem stāvokļiem, ko pavada periodisks vājums un mioglobinūrija. Karnitīna palmitiltransferāzes aktivitātes noteikšanas metodei ir diferenciāldiagnostikas vērtība. Tas samazinās muskuļos un aknu audi, leikocītu un fibroblastu kultūra. Diētas ievērošana, kas sastāv no ogļhidrātiem bagātinātiem un zema tauku satura pārtikas produktiem, palīdz samazināt uzbrukumu skaitu.
Muskuļu distrofijas. Šīs anomālijas pieder pie ģimenes slimību grupas, ko pavada muskuļu šķiedru deģenerācija. Muskuļu distrofiju klasifikācija balstās uz tādām pazīmēm kā sākuma laiks, progresēšanas ātrums, bojājumu sadalījums pa muskuļu grupām un mantojuma veids.
Pseidohipertrofiska muskuļu distrofija. Bērnība jeb Duchenne ir visizplatītākā muskuļu distrofijas forma; tā biežums ir 0,14 uz 1000 bērniem. Klasiskā formā tas notiek tikai zēniem, un ar X saistīta mantojums notiek aptuveni 50% probandu. Citos gadījumos slimību izraisa jaunas mutācijas. Ir ziņots par retu muskuļu distrofijas formu, kas klīniski ir identiska Dišēna formai, bet pārmantota recesīvā veidā ar tādu pašu slimības biežumu zēniem un meitenēm. Reti ir iespējams ticami diagnosticēt slimību bērnam līdz 3 gadu vecumam. Anamnēze parasti norāda, ka bērnam bija attīstības kavēšanās motoriskās funkcijas, viņš sāka vēlu sēdēt, staigāt un skriet, kas, protams, liecina par vairāk agrs sākums slimības. Izplatīta gaita, apgrūtināta kāpšana pa kāpnēm, ikru muskuļu hipertrofija ir izplatīta parādība klīniskās izpausmes. Dažos gadījumos procesā tiek iesaistīti arī citi muskuļi, jo īpaši deltveida, brahioradiālie un mēles muskuļi.
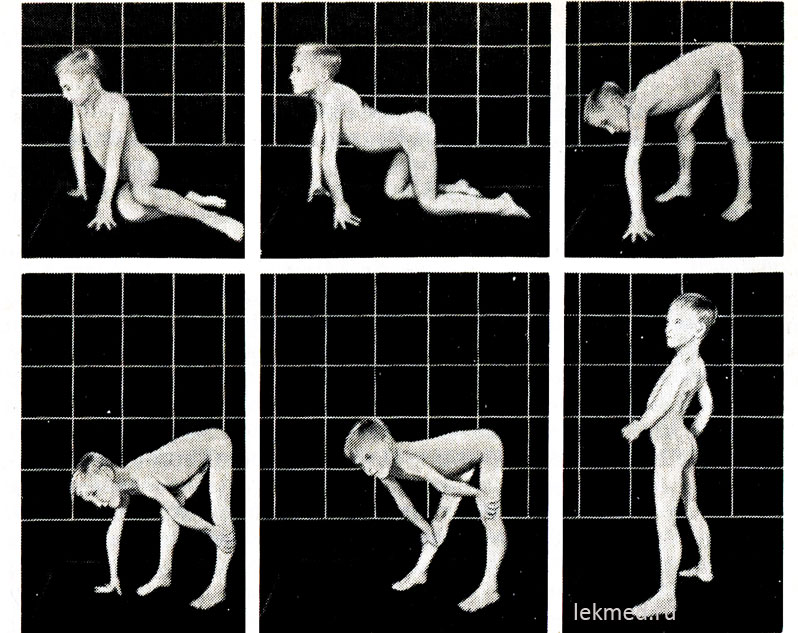
Rīsi. 21-9. Tipiskas pozas, ko ieņem 7 gadus vecs bērns ar pseidohipertrofisku miopātiju, paceļoties no grīdas (Gowersa zīme).
Stāv stāvoklī (pēdējā foto) ir ievērojami izteikta lordoze.
Slimības sākumā hipertrofētiem muskuļiem ir ievērojams spēks, bet vēlāk tas samazinās (pseidohipertrofija), jo muskuļu masas palielināšanās notiek to tauku infiltrācijas dēļ. Hipertrofētā ikru muskuļa spēks ievērojami pārsniedz kājas priekšējās virsmas muskuļu spēku, kas izskaidro biežas papēža cīpslas kontraktūras un bērna staigāšanu uz pirkstiem. Iegurņa jostas muskuļu vājums izpaužas raksturīgā pīlei līdzīgā (kundzīgā) gaitā un grūtībās, ko bērns izjūt, pieceļoties no sēdus stāvokļa uz grīdas. Diezgan smagās muskuļu distrofijas formās bērnam ir Goversa simptoms: pieceļoties no grīdas, viņš vispirms nometas ceļos, atspiedies uz rokām, un tad ceļas, secīgi nospiežot rokas no apakšstilbiem, ceļa locītavas un gurniem (21.-9. att.). Plecu jostas muskuļu vājumu var noteikt, turot bērnu paceltā stāvoklī aiz padusēm. Parasti viņš cenšas noturēties, piespiežot rokas pie ķermeņa; ar muskuļu distrofiju šķiet, ka tas izslīd cauri pārbaudītāja rokām. Slims bērns bieži nevar pacelt rokas virs galvas. Vēlākajos slimības posmos attīstās ievērojama muskuļu atrofija. Parasti līdz 12 gadu vecumam bērns vairs nevar staigāt. 75% gadījumu pacienti mirst pirms 20 gadu vecuma. Lielākajai daļai no viņiem ir kardiomiopātija, kas dažos gadījumos izraisa pēkšņa nāve. Ja mantojums ir saistīts ar X hromosomu un slimība sākās vēlā bērnībā, dzīves ilgums saglabājas garš (Becker muskuļu distrofija). Vidējais IQ bērniem ar Dišēnu ir 80; 25% bērnu ir garīga atpalicība.
Plkst diferenciāldiagnoze Jāņem vērā Dišena muskuļu distrofija vecākiem zīdaiņiem un muskuļu slimības, piemēram, endokrīnās miopātijas, karnitīna deficīts, glikogēna uzkrāšanās slimības un polimiozīts. Dažreiz ar papēža cīpslas kontraktūrām un bērna staigāšanu uz pirkstiem var pieņemt cerebrālo trieku, bet ar muskuļu distrofiju nav raksturīgu pazīmju. cerebrālā trieka spasticitāte un hiperrefleksija.
Diagnoze balstās uz seruma enzīmu aktivitātes noteikšanu, elektromiogrāfijas datiem un muskuļu audu biopsiju. Fermentu aktivitāte, īpaši kreatīnfosfokināze, pat pirms attīstības klīniskie simptomi pat zīdaiņiem bieži pārsniedz normu 10 reizes. Elektromiogramma galvenokārt atklāj motora potenciālu ilguma samazināšanos un amplitūdas samazināšanos. Histoloģiskās izmaiņas sastāv no muskuļu šķiedru deģenerācijas. Tie bieži atšķiras pēc izmēra un daļēji tiek aizstāti ar taukiem un saistaudiem. Arī to kodolu lielums ir atšķirīgs. Diagnozi var veikt dzimšanas brīdī, mērot kreatīnfosfokināzes aktivitāti. Sieviešu nēsātāju identificēšanas metodes vēl nav izstrādātas, neskatoties uz to, ka 60-80% no tiem ir neliels vai mērens tā līmeņa pieaugums. Šīs pazīmes ir vairāk raksturīgas bērnībai nekā nākamajiem dzīves periodiem.
Efektīvas metodes nav ārstēšanas. Pacientam jābūt aktīvam un jāspēj pēc iespējas vairāk staigāt. Ir jānodrošina, lai bērns izvairītos no intensīvas fiziskās slodzes, jo tas var izraisīt muskuļu šķiedru plīsumu. Dažos gadījumos ķirurģiska papēža cīpslas pagarināšana uzlabo spēju staigāt, bet ilgstoši gultas režīms pēc ortopēdiskās korekcijas var palielināties muskuļu atrofija. Ģenētiskajai konsultācijai ir svarīga loma.
Iedzimta muskuļu distrofija. Slimība ir iedzimta autosomāli recesīvā veidā, un tai raksturīga muskuļu hipotonija un vājums zīdaiņiem. Tas ir iekļauts stāvokļu grupā, kas definēts kā “vēss bērns” (sk. 21-1. tabulu). Slimības sākums ir pirmsdzemdību periodā. Dažreiz jaundzimušajam ir smaga muskuļu atrofija, kontraktūras un ierobežota locītavu kustīgums. Ir grūti atšķirt no Werdnig-Hoffmann slimības. Mēles fascikulācijas, kas raksturīgas pēdējai, muskuļu distrofijas gadījumā nav. Cīpslu refleksi ir nomākti, bet nav pilnībā zaudēti. Procesā tiek iesaistīti elpošanā iesaistītie muskuļi, tostarp diafragma. Smagos gadījumos nāve iestājas pirms 1 gada vecuma elpošanas mazspējas dēļ; vieglākās formās normāla dzīvotspēja saglabājas ilgu laiku. Seruma enzīmu aktivitātes palielināšanās netiek novērota, lai gan muskuļos notiek distrofiskas izmaiņas.
Muskuļu distrofijas sejas-humerālā forma. Pietiek ar šo viegla forma muskuļu distrofija tiek mantota autosomāli dominējošā veidā. Tas parasti sākas 10-20 gadu vecumā, un tam raksturīgs sejas un plecu jostas muskuļu vājums un atrofija. Seja ir pilnīgi draudzīga, pacients nevar aizvērt acis un svilpt. Slimība progresē lēni un ir saderīga ar normālu dzīves ilgumu. Diagnoze balstās uz klīniskajiem atklājumiem un mantojuma veidu. Muskuļu audu biopsijas rezultāti liecina par distrofiskām izmaiņām tajā. Kreatīnfosfokināzes līmenis serumā var palikt normas robežās vai var būt nedaudz paaugstināts.
Muskuļu distrofijas iegurņa forma. Šai neviendabīgo traucējumu grupai raksturīga lēna muskuļu distrofijas progresēšana, un tā tiek mantota autosomāli recesīvā veidā. Slimības sākums attiecas uz vecāku bērnību, pusaudžu vai pieaugušo vecumu. Parasti tiek ietekmēti iegurņa jostas muskuļi.
Acu miopātijas forma. Distrofiskas izmaiņas notiek galvenokārt ārējos acu muskuļos. Slimība sākas bērnībā vai pusaudža gados. Līdz ar to progresē ptoze un kustību ierobežojumi acs āboli. Dažreiz vājums izplatās uz sejas un kakla muskuļiem. Slimība ir jānošķir no myasthenia gravis un paralīzes galvaskausa nervi smadzeņu stumbra audzējiem.
Progresējoša oftalmopleģija, kas sākas bērnībā vai pusaudža gados, ir saistīta ar netipisku pigmenta tīklenes deģenerāciju un sirds blokādi (Kērnsa-Saijera sindroms). Tas parasti ir saistīts ar progresējošu ataksiju, augšanas aizkavēšanos un pubertāti. Zem muskuļu sarkolemmas tiek konstatēti lieli netipisku mitohondriju uzkrājumi. Šī procesa ģenētiskais raksturs nav noskaidrots. Pēkšņas nāves iespējamību no sirds vadīšanas problēmām var kontrolēt, izmantojot elektrokardiostimulatoru.
Miotoniskā distrofija. Neskatoties uz to, ka miotoniskā distrofija sākas it kā pieaugušam cilvēkam, tās sākums arvien biežāk tiek reģistrēts zīdaiņiem un vēlāk bērnībā. Tas tiek mantots autosomāli dominējošā veidā. Tās parādīšanās bērnībā liecina, ka māte cieš no miotonijas. Attiecīgi intrauterīni faktori var ietekmēt bērna slimības smagumu. Jau dzimšanas brīdī viņu var noteikt muskuļu hipotonija, viņam trūkst sūkšanas spējas. Fiziskās un garīgās attīstības aizkavēšanās parasti tiek atklāta vēlāk. Agrā bērnībā muskuļu vājums un atrofija izplatījās galvenokārt uz sejas, žokļa un pagaidu muskuļiem. Parasti tiek atzīmēta divpusēja ptoze. Diagnostiski nozīmīgas metodes ietver muskuļu perkusiju, elektromiogrāfiju; tipiska šiem pacientiem ir nespēja atspiest roku, kas savilkta dūrē (sk. Iedzimta miotonija). Ekstremitāšu un iegurņa jostas (parasti distālās grupas) muskuļu vājums un atrofija tiek konstatēta vecākā bērnībā vai pusaudža gados. Pieaugušajiem šo slimību pavada katarakta, baldness un sēklinieku atrofija.
Diagnoze balstās uz miotonijas pazīmju identificēšanu, raksturīgo muskuļu vājuma sadalījumu, dominējošo iedzimtību un distrofiskām izmaiņām muskuļos. Bērnībā slimības gaita var būt nelabvēlīga, un to bieži pavada garīga atpalicība. Pusaudža gados priekšplānā izvirzās muskuļu vājums. Funkcionālo traucējumu gadījumā ir indicēta ārstēšana ar novokaīnu un hinidīnu.

