Amyotrofinen lateraaliskleroosi (ALS), joka tunnetaan myös nimellä Lou Gehrigin tauti ja Charcotin tauti, tietty sairaus joka johtaa solukuolemaan. Isossa-Britanniassa käytetään termiä motorinen neuronitauti (MND), kun taas muissa maissa termiä käytetään viiteen tyyppiseen sairauteen, joista ALS on yleisin. Sille on ominaista lihasten surkastuminen, lihaskrampit ja vähitellen lisääntyvä heikkous, joka johtuu lihasten menetyksestä lihaskudos. Tämä johtaa puhe-, nielemis- ja joissakin tapauksissa hengitysvaikeuksiin. Syy on tuntematon 90-95 prosentissa tapauksista. Noin 5-10 % tapauksista on perinnöllisiä. Noin puolet tapauksista johtuu yhdestä tai kahdesta spesifisestä geenistä. Tämä johtaa tahallista lihasten supistumista säätelevien neuronien kuolemaan. Diagnoosi perustuu merkkeihin ja oireisiin testauksen jälkeen muiden mahdollisten sairauksien sulkemiseksi pois. ALS:iin ei ole parannuskeinoa. Rilutsoli-niminen lääke hidastaa ALS:n etenemistä ja pidentää elinikää 2-3 kuukaudella. Non-invasiivinen ventilaatio voi parantaa potilaan tilaa ja elinikää. Sairaus kehittyy yleensä 60 vuoden iästä alkaen, jos kyseessä perinnöllinen sairaus– alkaen 50. Keskimääräinen elinajanodote taudin alkamisesta on 3-4 vuotta. Noin 10 % potilaista elää yli 10 vuotta. Suurin osa kuolee hengityskatkoihin. Useimmissa maissa ALS:n ilmaantuvuutta ei tunneta. Euroopassa ja Yhdysvalloissa tautia esiintyy kahdella 100 000 ihmisestä vuodessa. Charles Bell kuvasi taudin ensimmäisen kerran vuonna 1824. Jean-Martin Charcot kuvasi oireiden ja taustalla olevien neurologisten ongelmien välisen suhteen vuonna 1869, ja vuonna 1874 hän alkoi käyttää termiä "amyotrofinen lateraaliskleroosi". Yhdysvalloissa tauti tuli tunnetuksi sen jälkeen, kun se osui kuuluisaan baseball-pelaaja Lou Gehrigiin, ja Stephen Hawking tuli tunnetuksi tieteellisistä saavutuksistaan. Vuonna 2014 käynnistettiin verkkokampanja ALS-tietoisuuden lisäämiseksi. Osana tätä jääsämpärihaasteeksi kutsuttua kampanjaa (kirjaimellisesti - testi jäävesiämpärillä) ihmiset kastelivat itseään vesiämpärillä ja tekivät lahjoituksia.
Merkit ja oireet
Sairaus aiheuttaa lihasten heikkoutta ja tuhlausta koko kehossa ylempien ja alempien motoristen neuronien rappeutumisen vuoksi. Sairastuneet voivat lopulta menettää kyvyn hallita vapaaehtoisia liikkeitä, mutta virtsarakko, suolet ja silmien liikelihakset toimivat taudin pitkälle edenneeseen vaiheeseen asti. Myös kognitiivinen toiminta säilyy useimmilla ihmisillä, vaikka jotkut (noin 5 %) kehittävät frontotemporaalista dementiaa. Monet (30–50 %) kokevat myös pieniä kognitiivisia muutoksia, jotka saattavat jäädä huomaamatta, mutta jotka näkyvät yksityiskohtaisessa neuropsykologisessa testauksessa. ALS-tautia havaitaan harvoin ihmisillä, joilla on rappeuttava lihas- ja luusairaus (osa monisysteemisen proteinopatian oireyhtymää). Sairaus ei vaikuta aistihermoihin ja autonomiseen hermostoon, mikä tarkoittaa, että useimmat ihmiset säilyttävät kuulonsa, näkönsä, herkkyytensä, hajunsa ja makunsa.
Ensisijaiset oireet
Taudin varhaiset oireet ovat melko yleisiä (heikkous ja/tai lihasten surkastuminen), joten diagnoosi on vaikeaa. Muita oireita ovat nielemisvaikeudet, sairastuneiden lihasten kouristukset tai jäykkyys, lihasheikkous (käsissä tai jaloissa) ja/tai epäselvä puhe ja nenäkäytävät. Taudin ensimmäisissä vaiheissa se kehon osa vaikuttaa sen mukaan, mitkä motoriset neuronit vaurioituvat ensin. Noin 75 % sairastavista tuntee ensin käsien tai jalkojen heikkoutta tai surkastumista. Kävelyssä tai juostessa esiintyy kömpelyyttä, henkilö voi kompastua tai kompastua tai vetää jalkaansa hieman takanaan (jalan pudotusoireyhtymä). Jos käsi vaikuttaa, käden taitoa vaativissa liikkeissä on vaikeuksia (esimerkiksi paidan nappiminen, kirjoittaminen, avaimen kääntäminen lukossa). Oireet voivat jatkua vain yhdessä raajassa pitkän aikaa tai koko sairauden ajan; oire tunnetaan yhden raajan amyotrofiana. Noin 25 % kaikista sairauksista alkaa etenevänä bulbar-oireyhtymänä. Ensisijaiset oireet ilmenevät tässä tapauksessa puhe- tai nielemisvaikeuksina. Puhe muuttuu sekavammaksi, hiljaisemmaksi ja nasaaliseksi. Nielemisvaikeuksiin liittyy kielen liikkuvuuden menetys. Pienellä määrällä ihmisiä hengitysprosessiin vaikuttavat kylkiluiden väliset lihakset kärsivät ensimmäisinä. Pienellä osalla ihmisistä on frontotemporaalinen dementia, mutta taudin edetessä ilmenee tyypillisempiä ALS-oireita. Ajan myötä ihmisillä on vaikeuksia liikkua, niellä (dysfagia), puhua ja muodostaa sanoja (dysartria). Ylempien motoristen hermosolujen toimintahäiriö ilmenee lihasten kovuuden (spastisuuden) ja lisääntyneenä refleksitoimintaa(hyperrefleksia) sekä hyperaktiivinen gag-refleksi. Babinskin refleksi osoittaa myös ylemmän motorisen neuronin vaurion. Alemman motorisen hermosolun vaurion oireita ovat lihasheikkous ja surkastuminen, ihon alla näkyvät lihaskrampit, mutta pienet kouristukset eivät ole diagnosoitavissa oleva oire, ne havaitaan myöhemmin tai liittyvät heikkouteen ja atrofiaan. Noin 15–45 % ihmisistä kokee affektiivista labiliteettia, neurologista häiriötä, joka tunnetaan emotionaalisena epävakauteena ja joka koostuu hallitsemattomasta naurusta, itkusta ja jatkuvasta hymyilystä, joka liittyy sipulin ylempien motoristen neuronien rappeutumiseen, joka ilmenee liiallisessa tunteiden ilmaisussa. . ALS-diagnoosin saamiseksi henkilöllä on oltava ylemmän ja alemman hermosolujen vaurion oireita, jotka eivät liity muihin sairauksiin.
Taudin kehittyminen
Vaikka oireiden kehittymisjärjestys ja -nopeus vaihtelee yksilöiden välillä, useimmat ihmiset eivät pysty kävelemään tai käyttämään käsiään. He menettävät myös kyvyn puhua ja niellä ruokaa, joten BiPAP ei-invasiivista ventilaatiota (BiPAP) käytetään yleisimmin. Taudin etenemisnopeutta arvioidaan ALSFRS-R-asteikolla, joka on kyselylomake tai kliininen haastattelu asteikolla 48 (normaali toiminta) 0:aan (vaikea muoto). Vaikka vaihteluaste on suuri ja pienellä osalla ihmisistä sairausaste on alhainen, potilaat menettävät keskimäärin 0,9 pistettä kuukaudessa. Kliinisiin tutkimuksiin perustuva tutkimus osoitti 20 %:n muutoksen ALSFRS-R:ssä kliinisesti merkittävänä. Riippumatta siitä, mihin kehon osaan ensin vaikuttaa, kehittyvä sairaus leviää muihin kehon osiin. Raajojen oireet leviävät yleensä vastakkaisiin raajoihin eikä uuteen kehon osaan, kun taas bulbar-sairaus vaikuttaa ensin käsiin ja sitten jalkoihin. Taudin etenemisnopeus on alhaisempi potilailla, jotka ovat alle 40-vuotiaita taudin alkaessa, potilailla, joilla on alhainen liikalihavuus, henkilöillä, joilla on sairaus toisessa raajassa, ja niillä, joilla on ylemmän motorisen hermosolun sairauden primaarisia oireita. Sitä vastoin taudin etenemisnopeus on korkeampi ja ennuste huonompi ihmisillä, joilla on bulbar-, hengitystie- ja frontotemporaalinen dementia. CX3CR1-alleelisella variantilla on vaikutusta taudin etenemiseen ja elinajanodotteeseen.
Myöhäiset vaiheet
Vaikka avustettu ventilaatio voi lievittää hengitysongelmia ja pidentää elinikää, se ei hidasta ALS:n etenemistä. Suurin osa ALS-potilaista kuolee hengityspysähdyksissä 3–5 vuoden kuluessa oireiden alkamisesta. Keskimääräinen elinajanodote taudin puhkeamisesta kuolemaan on 39 kuukautta, ja vain 4 % ihmisistä elää yli 10 vuotta. Kitaristi Jason Becker on elänyt taudin kanssa vuodesta 1989, ja fyysikko Stephen Hawking on elänyt taudin kanssa yli 50 vuotta, mutta nämä tapaukset ovat kaikki ainutlaatuisia. Pureskelu- ja nielemisvaikeudet vaikeuttavat syömistä ja lisäävät tukehtumisriskiä ja ruoan nielemistä keuhkoihin. Sairauden myöhemmissä vaiheissa voi kehittyä aspiraatiokeuhkokuume ja painonpidosta voi tulla vakava ongelma, joka voi vaatia syöttöletkun. Pallean ja kylkiluiden välisten lihasten heikkenemisen kanssa rinnassa, jotka tukevat hengitystä, keuhkojen toimintaa tai pikemminkin elinkykyä ja sisäänhengityspainetta laskevat. Sairauden hengitystiemuodossa tämä voi tapahtua myös ennen raajojen lihasten heikkenemistä. Suurin osa ALS-potilaista kuolee hengityspysähdykseen tai keuhkokuumeeseen. Päällä viimeiset vaiheet sairauteen voi vaikuttaa okulomotorinen hermo, joka ohjaa liikettä, kuten silmämunan lihaksia. Silmien liike säilyy viimeisiin vaiheisiin asti lihaserojen vuoksi silmämuna luustolihasten kanssa, joihin tauti vaikuttaa ensimmäisinä. Sairauden myöhemmissä vaiheissa potilaan tila voi muistuttaa locked-in-oireyhtymää.
silmien liikettä
ALS-potilailla voi olla vaikeuksia tehdä nopeita vapaaehtoisia silmän liikkeitä. Silmien liikenopeus hidastuu. Siellä on myös silmien lähentymiskouristuksia. Vestibulo-silmän refleksin testaus voi auttaa tunnistamaan nämä ongelmat. Elektrookulografia (EOG) mittaa verkkokalvon lepopotentiaalia. ALS-potilailla EOG osoittaa muutoksia, jotka vaikuttavat taudin etenemiseen, ja tarjoaa myös tietoa silmän motoriseen toimintaan vaikuttavan taudin etenemisen kliiniseen arviointiin. Lisäksi EOG:n avulla voit tunnistaa silmäongelmat varhaisessa vaiheessa. Silmänmotoriset lihakset eroavat muista luustolihaksista. Silmänmotorinen lihas on ainutlaatuinen siinä mielessä, että se muuttuu jatkuvasti koko elämän ajan ja ylläpitää satelliittisolujen määrää ikääntymisen aikana. Silmänmotorisissa lihaksissa on paljon enemmän myogeenisiä progenitorisoluja kuin raajojen luustolihaksissa.
Syyt
Genetiikka
5-10 %:ssa tapauksista sairaus periytyy suoraan vanhemmilta. Noin 20 % perhetapauksista (tai 2 % kaikista tapauksista) liittyy mutaatioon kromosomissa 21, joka koodaa superoksididismutaasia. Mutaatiomuotoja on yli sata. SISÄÄN Pohjois-Amerikka yleisin mutanttigeeni on mutantti SOD1-geeni; sille on ominaista uskomattoman nopea etenemisnopeus sairauden hetkestä kuolemaan. Skandinavian maiden yleisin mutantti on D90A-SOD1, jonka etenemisnopeus on hitaampi kuin tyypillinen ALS, ja tätä sairautta sairastavat ihmiset elävät keskimäärin 11 vuotta. Vuonna 2011 C9orf72:ssa löydettiin heksanukleotiditoistona tunnettu geneettinen poikkeama, joka liittyy ALS:ään ja frontotemporaaliseen dementiaan, ja se on jopa 6 % kaikista valkoisten eurooppalaisten ALS-tapauksista. Tämä geeni esiintyy myös filippiiniläisistä ihmisistä. UBQLN2-geeni vastaa ubiquilin 2 -proteiinin tuotannosta solussa, joka on ubikiliiniperheen jäsen ja säätelee ubiquitinoitujen proteiinien hajoamista. Mutaatiot UBQLN2:ssa estävät proteiinien hajoamista, mikä johtaa hermosolujen rappeutumiseen ja aiheuttaa (pääasiassa perinnöllistä) X-kytkettyä ALS:ää ja ALS/dementiaa.
SOD1
Vuonna 1993 tutkijat havaitsivat, että geenin (SOD1) mutaatio, joka tuottaa Cu-Zn-superoksididismutaasin (SOD1) entsyymiä, liittyy 20 prosenttiin perinnöllisistä ALS-tapauksista. Tämä entsyymi on melko voimakas antioksidantti, joka suojaa kehoa superoksidin, myrkyllisen vapaan radikaalin, aiheuttamilta mitokondrioissa syntyviltä vaurioilta. Vapaat radikaalit ovat erittäin reaktiivisia molekyylejä, joita solut tuottavat aineenvaihdunnan aikana. Vapaat radikaalit voivat kerääntyä ja vahingoittaa DNA:ta ja proteiineja solussa. Tällä hetkellä yli 110 erilaista mutaatiota SOD1:ssä liittyy ALS:ään, joista joillakin (kuten H46R) on erittäin pitkä kliininen historia, kun taas toisilla, kuten A4V, on poikkeuksellisen aggressiivisia. Kun puolustus oksidatiivista stressiä vastaan heikkenee, solukuolema (apoptoosi) aktivoituu. Vika SOD1:ssä voi johtaa toiminnan menetykseen tai lisääntymiseen. SOD1-toiminnan menetys voi johtaa vapaiden radikaalien kerääntymiseen. SOD1-toimintojen hankkiminen voi olla myrkyllistä. Siirtogeenisiä hiiriä koskevien tutkimusten tuloksena on esitetty useita teorioita SOD1:n roolista perinnöllisissä ALS:issä. Hiiret, joilta puuttuu SOD1-geeni, eivät kehitä ALS:ää, vaikka he kokevatkin nopeutunutta ikääntymiseen liittyvää lihasatrofiaa (sarkopenia) ja lyhentynyttä elinajanodotetta. Tämä tarkoittaa, että SOD1-mutaation myrkylliset ominaisuudet ovat seurausta toiminnan lisääntymisestä, eivät menetyksestä. Lisäksi proteiinien kertymisen on havaittu olevan perinnöllisen ja satunnaisen ALS:n (proteinopatian) patologinen piirre. Kummallista kyllä, hiirissä, joilla oli mutatoitunut SOD1 (yleisimmin G93A-mutaatio), mutatoidun SOD1:n kertymistä (väärin laskostunut proteiini) havaittiin vain sairastuneissa kudoksissa, ja suurempi kertyminen havaittiin motoristen hermosolujen rappeutumisen aikana. Tutkijat uskovat, että mutatoituneen SOD1:n kertymisellä on tärkeä rooli solujen toiminnan häiriintymisessä mitokondrioiden, proteasomien, kaperonien ja muiden proteiinien vaurioitumisen kautta. Jos ne vahvistetaan, mikä tahansa näistä poikkeavuuksista voi toimia todisteena siitä, että tällaiset kertymät johtavat mutatoidun mutatoidun SOD1:n toksisuuteen. Kriitikot huomauttavat, että ihmisillä SOD1-mutaatiot aiheuttavat vain 2 % kaikista sairauksista, ja syy-mekanismit voivat poiketa taudin satunnaisesta muodosta vastuussa olevista. Tällä hetkellä ALS-SOD1-hiiret ovat edelleen paras malli taudista prekliinisissä tutkimuksissa, mutta toivotaan, että uusi malli kehitetään. Käytettävissä on online-tietokanta, joka on tieteellinen yhteisö ja foorumi, joka sisältää ajan tasalla olevaa ALS-tietoa suurelle yleisölle. Sivusto on nimeltään ALSOD, se luotiin alun perin SOD1-geeniä koskevia julkaisuja varten vuonna 1999, tällä hetkellä sivustolla on yli 40 ALS-geeniin liittyvää geeniä.
Muut tekijät
Jos tauti ei ole perinnöllinen, eli 90 %:ssa tapauksista, taudin syitä ei tunneta. Mahdollisia syitä vaikka epäluotettavia, ovat päävammat, asepalvelus, toistuva huumeiden käyttö ja osallistuminen kontaktiurheiluun. Tutkimus keskittyy myös glutamaatin rooliin motoristen hermosolujen rappeutumisessa. Glutamaatti on yksi aivojen välittäjäaineista. Tutkijat ovat havainneet, että ALS-potilailla on enemmän terveisiin verrattuna korkeatasoinen glutamaatti veressä ja aivo-selkäydinnesteessä. Rilutsoli on tällä hetkellä ainoa Yhdysvalloissa hyväksytty lääke ALS:n hoitoon ja vaikutuksiin glutamaatin kuljettajiin. Lääkkeellä on alhainen vaikutus eliniän pidentämiseen, mikä viittaa siihen, että liiallinen glutamaatti ei ole taudin ainoa syy. Jotkut tutkimukset viittaavat siihen, että satunnaisen ALS:n (etenkin urheilijoilla) ja ruokavalion, joka sisältää runsaasti haaraketjuisia aminohappoja (suosittu lisäravinne urheilijoiden keskuudessa), välillä on yhteys. Solujen kiihtyvyys johtaa lisääntyneeseen kalsiumin imeytymiseen solussa, mikä johtaa hermosolujen kuolemaan. Jotkut todisteet viittaavat siihen, että superoksididismutaasi 1 (SOD1) -proteiinin rakenteen muodostumisen pervasiivinen häiriö tapahtuu samalla tavalla kuin prioneissa. β-metyyliamino-l-alaniinin (BMAA) sisällyttämisen oletetaan myös johtavan toiseen prionien kaltaiseen epäjärjestyneen proteiinirakenteen muodostumisen leviämiseen. Toinen yleinen ALS:ään liittyvä tekijä on leesio moottorijärjestelmä alueilla, kuten frontotemporaalisissa lohkoissa. Tappio tällä alueella on merkki varhaiset rikkomukset, jota voidaan käyttää tappion ennustamiseen moottoritoiminto. ALS:n mekanismit ilmenevät kauan ennen kuin ensimmäiset merkit ja oireet ilmaantuvat. Ennen kuin lihasatrofia tulee ilmeiseksi, noin kolmanneksen motorisista neuroneista täytyy kuolla. Monia muita mahdollisia riskitekijöitä on tutkittu - altistuminen kemikaaleille, sähkömagneettisille kentille, fyysinen loukkaantuminen ja sähköisku - mutta johdonmukaisia johtopäätöksiä ei ole tehty.
Patofysiologia
ALS:n erottuva piirre on ylempien ja alempien motoristen neuronien kuolema aivokuoren, aivorungon ja selkäytimen projektioalueella. Ennen kuolemaansa motoriset neuronit kehittävät proteiinipitoisia sulkeumia solurungossa ja aksoneissa. Osittain tämä voi johtua proteiinien hajoamisesta. Nämä sulkeumat sisältävät usein ubikvitiinia ja sisältävät usein yhden ALS-proteiineista: SOD1, TAR DNA:ta sitova proteiini (TDP-43 tai TARDBP) tai FUS RNA:ta sitova proteiini.
Luuston motoriset yksiköt
Silmämunan ulkoiset lihakset ja luustolihakset näyttää erilaisia ominaisuuksia. Alla on lueteltu ominaisuuksia, jotka erottavat silmämunan lihakset luurankolihaksista.
Yksi hermosäike liittyy vain yhteen lihaskuituun
Ei venytysrefleksiä huolimatta suuri määrä lihaskarat
Ei syklistä estoa
Nopeiden/hitaiden lihasten puute
Kaikki silmän motoriset neuronit osallistuvat kaikentyyppisiin silmän liikkeisiin.
Erot havaitaan myös terveiden ja sairaiden silmämunan lihasten välillä. Kuolleiden luovuttajien silmämunan lihakset säilyttävät sytoarkkitehtoninsa verrattuna raajojen lihaksiin. Terveet silmämunan lihakset koostuvat silmämunan edessä olevasta keskikerroksesta (GL) ja silmäkulman edessä olevasta ohuesta orbitaalikerroksesta (OL). Silmänmotoriset lihakset, joihin ALS vaikuttaa, säilyttävät GL:n ja OL:n asennon. Silmänmotoriset lihakset säilyttävät aivoista peräisin olevan neurotrofisen tekijän (BDNF) ja glialisen neurotrofisen tekijän, jotka myös säilyvät ALS:n saaneissa lihaksissa. Laminiini on rakenteellinen proteiini, joka sijaitsee neuromuskulaarisessa liitoksessa (NMJ). Lnα4 on laminiinin isomuoto, joka on tunnusmerkki silmän motoristen lihasten neuromuskulaarinen liitos. Ihmiset, joilla on ALS, säilyttävät Lnα4:n ilmentymisen hermo-lihaksen okulomotorisessa liitoksessa, mutta tämä ilmentymä puuttuu samojen ihmisten raajojen lihaksista. Laminiinin ilmentymisen ylläpitämisellä voi olla tärkeä rooli silmän motoristen lihasten eheyden ylläpitämisessä ALS-potilailla. Ihmisillä, joilla on satunnainen ALS (sALS), on kohonnut taso solunsisäinen kalsium, joka lisää välittäjäaineiden vapautumista. Passiivinen seerumin kuljetus ihmisiltä, joilla on sALS, lisää välittäjien spontaania vapautumista aivo-selkäydinnesteessä. Silmänmotoriset lihakset vastustavat fysiologisen tilan muutoksia. Kuitenkin on erilaisia taudin vaikutus. ALS:sta kärsivillä silmämotorisilla lihaksilla on suurempi vaihtelu säikeiden koossa kuin terveillä kontrollilihaksilla. Silmämotorisista lihaksista löydettiin klusteroituneita ja hajallaan olevia atrofisia ja hypertrofisia säikeitä, mutta näiden lihasten vauriot ovat huomattavasti pienempiä kuin samojen luovuttajien raajojen lihaksissa. Myös sidekudos ja rasvakudos lisääntyvät silmän motorisissa lihaksissa kuitujen katoamisen ja surkastumisen kompensoimiseksi. ALS-potilailla on myös oftalmoplegia, hermosolujen menetys silmämunan motoristen lihasten ytimien ympärillä ja sisällä. Lisäksi myosiinin raskaan ketjun pitoisuus silmän motoristen lihasten säikeissä muuttuu, hitaan myosiinin raskaan ketjun normaali ilmentyminen GL:ssä häiriintyy ja alkion myosiinin raskas ketju puuttuu OL:sta. Muutokset hitaan myosiinin raskaassa ketjussa ja alkion myosiinin raskaassa ketjussa ovat ainoat muutokset silmän motorisissa lihaksissa. Koska okulomotorinen lihas on erittäin hermottunut, kaikki denervaatiot kompensoidaan viereisillä asconeilla, jotka pysyvät toiminnassa.
laktaatti ja cinnamaatti
Maitohappo - lopputuote glykolyysi, joka aiheuttaa lihasten väsymistä. Laktaattidehydrogenaasientsyymi toimii kaksisuuntaisesti ja voi hapettaa laktaatin pyruvaatiksi, joten sitä voidaan käyttää Krebsin syklissä. Silmänmotorisissa lihaksissa laktaatti tukee lihasten supistumista lisääntyneen aktiivisuuden aikana. Uskotaan, että okulomotoriset lihakset korkea aktiivisuus Laktaattidehydrogenaasit ovat resistenttejä ALS:lle. Cinnamate on laktaatin kuljetuksen estäjä. Cinnamate voi väsyttää glosomotorisia lihaksia vähentäen lihasten kestävyyttä ja jäännösponnistusta. Sinnamaatilla ei kuitenkaan ole vaikutusta varpaan ojentajalihaksiin. Glukoosin korvaaminen eksogeenisellä laktaatilla lisää digitorum longus -lihasten väsymistä, mutta sillä ei ole vaikutusta glosomotoriseen lihakseen. Silmämotoristen lihasten väsymistä havaitaan vain, kun eksogeenisen laktaatin ja sinamaatin yhdistelmä korvaa glukoosin.
Diagnostiikka
Mikään analyysi ei pysty diagnosoimaan tarkasti ALS:ää, vaikka ylempien ja alempien motoristen hermosolujen kuolemaa osoittavien merkkien esiintyminen on olennainen merkki. ALS-diagnoosi tapahtuu ensisijaisesti lääkärin havaintojen ja muiden sairauksien poissulkevien testien perusteella. Lääkäri kerää potilaan täydellisen sairaushistorian ja suorittaa säännöllisesti neurologisia tutkimuksia oireiden, kuten heikkouden, lihasatrofian, hyperrefleksian ja lihasspassisuuden, etenemisen arvioimiseksi. Lisätestejä voidaan tehdä muiden sairauksien mahdollisuuden sulkemiseksi pois, koska monet ALS:n oireet voivat ilmaantua myös muiden hoidettavissa olevien sairauksien yhteydessä. Yksi tällainen testi on elektromyografia (EMG), joka havaitsee sähköisen toiminnan lihaksissa. Jotkut EMG-löydökset voivat tukea ALS-diagnoosia. Toinen yleinen testi on hermojohtonopeustesti (NVR). Testin tuloksena havaittu tietty poikkeama voi viitata siihen, että potilaalla on perifeerisen neuropatian muoto (vaurio ääreishermot) tai myopatia (lihassairaus), ei ALS. Magneettiresonanssikuvaus voi havaita poikkeavuuksia, jotka aiheuttavat ALS-oireita, kuten turvotusta selkäydin, multippeliskleroosi, kohdunkaulan välilevytyrä, syringomyelia tai kohdunkaulan spondyloosi. Oireiden ja testitulosten perusteella lääkärisi voi määrätä veri- ja virtsakokeita tai muita laboratoriotutkimuksia sulkeakseen pois muita mahdolliset sairaudet. Joissakin tapauksissa, jos lääkäri epäilee, että potilaalla on myopatia eikä ALS, hän voi myös tilata lihasbiopsian. Virussairaudet, kuten ihmisen immuunikatovirus (HIV), virus T-soluleukemia ihmisen (HTLS), Lymen taudin, kupan ja puutiaisaivotulehdus voi aiheuttaa ALS:n kaltaisia oireita. Neurologiset sairaudet kuten multippeliskleroosi, polion jälkeinen oireyhtymä, multifokaalinen motorinen neuropatia, Guillain-Barrén oireyhtymä ja spinaalinen lihasatrofia, voivat myös olla ALS:n kaltaisia. On tärkeää diagnosoida sairaus oikein, koska ALS:n oireet voidaan helposti sekoittaa useiden muiden sairauksien oireisiin. Siksi pätevän neurologin arviointi on tarpeen muiden sairauksien sulkemiseksi pois. Useimmissa tapauksissa ALS on helppo diagnosoida, ja virheelliset diagnoosit ovat alle 10 % tapauksista. Tutkimussuunnitelmalla ja säännöllisillä tutkimuksilla suoritettiin tutkimus, johon osallistui 190 MND/ALS-kriteerit täyttävää potilasta. 30 potilaan (16 %) diagnoosi muuttui dramaattisesti kliinisen seurantajakson aikana. Samassa tutkimuksessa kolmella potilaalla oli vääränlainen diagnoosi, myasthenia gravis (TM, autoimmuunisairaus). TM:llä on samat oireet kuin ALS:llä ja useilla muilla neurologisilla poikkeavuuksilla, mikä johtaa diagnoosin ja hoidon viivästymiseen. TM on hoidettavissa, mutta ALS ei. Myasteeninen oireyhtymä, muuten Lambert-Eatonin oireyhtymä, voi jäljitellä ALS:ää ja varhaiset oireet samanlainen kuin TM:n oireet.
Hoito
ALS-potilaille tarvitaan hoitoa oireiden lievittämiseksi ja eliniän pidentämiseksi. Monitieteinen lääketieteellinen tiimi, joka työskentelee ALS-potilaiden kanssa, uskoo, että tukihoito on välttämätöntä potilaiden pysymiselle aktiivisena ja mukavana.
Lääketieteelliset valmisteet
Rilutsoli (Rilutek) - lisää hieman potilaiden elinajanodotetta. Se pidentää elinikää useilla kuukausilla ja vaikuttaa enemmän potilaisiin, joilla on bulbar ALS. Lääkkeen avulla voit myös viivyttää ilmanvaihdon käyttöä. Lääkettä käyttäville potilaille on tehtävä maksatutkimus (10 % ihmisistä kärsii maksavauriosta ottamisen yhteydessä). Lääke on Yhdysvaltain terveysministeriön hyväksymä ja sitä suositellaan käytettäväksi. Kansallinen instituutti kliininen pätevyys. Rilutsoli ei korjaa motorisille neuroneille jo tehtyjä vaurioita. Muita lääkkeitä voidaan käyttää vähentämään väsymystä, helpottamaan lihaskramppeja, hallitsemaan spastisuutta ja vähentämään lisääntynyt syljeneritys ja ysköstä. Useat lääkkeet voivat myös lievittää kipua, masennusta, parantaa unta, vähentää dysfagiaa ja ummetusta. Baklofeenia ja diatsepaamia määrätään kontrolloimaan ALS-spastisuutta. Triheksifenidyyliä tai amitriptyliiniä voidaan määrätä, jos potilailla on vaikeuksia niellä sylkeä.
Hengitystuki
Kun hengitykseen osallistuvat lihakset ovat heikentyneet, voidaan tukea hengitystä ventilaatiolla (positiivinen paineventilaatio, kaksitasoinen positiivinen hengitysteiden paine (BiPAP) tai IVL-kaksivaiheinen ventilaatio (BCV)). Tällaiset laitteet pakottavat keuhkot keinotekoisesti toimimaan kasvoilla ja vartalolla olevien ulkoisten laitteiden avulla. Kun lihakset eivät enää pysty ylläpitämään happitasoa ja hiilidioksidi, näitä laitteita voidaan käyttää pysyvästi. BCV:llä on selkeä etu, että laite voi poistaa eritteet korkeataajuisilla värähtelyillä, joita esiintyy muutaman uloshengityksen jälkeen. Potilaat voivat myös harkita mekaanisen ventilaation (hengityksensuojainten) käyttöä, jolloin laite täyttää ja tyhjentää keuhkot. varten tehokas käyttö tarvitaan letku, jonka tulee kulkea nenästä tai suusta henkitorven läpi. Tällaisen laitteen pitkäaikainen käyttö voi vaatia leikkauksen, trakeotomia, jonka aikana putki työnnetään suoraan henkilön henkitorveen kaulassa olevan aukon kautta. Potilaiden ja heidän perheidensä tulee ottaa huomioon useita tekijöitä päättäessään, milloin käyttää jotakin yllä kuvatuista laitteista ja käyttääkö sitä ollenkaan. Tuulettimien vaikutus elämänlaatuun ja hinta vaihtelevat. Vaikka ventilaatio voi auttaa lievittämään hengitysongelmia ja siten pidentää elinikää, se ei vaikuta ALS:n etenemiseen. Ennen kuin he päättävät, minkä laitteen valita, potilaiden tulee olla täysin tietoisia niistä ja niiden vaikutuksista elämään ilman liikkumista. Jotkut ihmiset, joilla on pitkittynyt trakeotomia, ylipainehengitys erityisillä koneilla tai putkilla, voivat puhua, jos lihaksissa ei ole vaikutusta suuontelon(mutta joka tapauksessa taudin edetessä puhe katoaa). Muut potilaat voivat käyttää puheventtiiliä (kuten Passy-Mure-puheventtiiliä) puheterapeutin ohjauksessa. BiPAP-hengitystilassa toimivia ulkoisia ventilaattoreita käytetään ylläpitämään hengitystä ensin yöllä ja sitten päivällä. BiPAP:n käyttö on väliaikainen toimenpide. Kauan ennen kuin BiPAP ei ole enää tehokas, potilaiden tulisi harkita trakeotomiaa ja pitkäaikaista mekaanista ventilaatiota. Tässä vaiheessa jotkut potilaat valitsevat saattohoidon.
Terapia
Fysioterapialla on tärkeä rooli kuntoutusprosessissa, sillä on myönteinen vaikutus ALS-potilaisiin, mikä mahdollistaa voimanmenetyksen viivyttämisen, kestävyyden ylläpitämisen, kipujen vähentämisen, komplikaatioiden ehkäisyn ja toiminnan itsenäisyyden varmistamisen. Kuntoutusterapia ja erikoislaitteet auttavat myös varmistamaan potilaiden riippumattomuuden ja turvallisuuden ALS:n aikana. Kevyt aerobinen harjoittelu, kuten kävely, uinti ja pyöräily, vahvistaa koskemattomia lihaksia, parantaa sydän- ja verisuoniterveyttä ja auttaa potilaita selviytymään väsymyksestä ja masennuksesta. Vuorottelu- ja venytysharjoitukset ehkäisevät lihaskramppeja ja lihasten supistumista. Terapeutien on varmistettava, että lihakset ovat kuormitettuja ilman, että ne joutuvat ylikuormittamaan. Rampit, olkaimet, kävelijät, kylpyhuonetarvikkeet, pyörätuolit mikä auttaa potilaita pysymään liikkuvina. Terapeutit voivat suositella laitteita tai laitteita, jotka auttavat potilaita pysymään mahdollisimman turvallisena ja normaalina. Potilaat, joilla on puhevaikeuksia, voivat työskennellä puhevamma-asiantuntijoiden kanssa auttaakseen potilaita opettamaan tekniikoita, kuten puhumaan kovemmin ja selkeämmin. Sairauden edetessä asiantuntijat voivat suositella puhetta vahvistavien laitteiden käyttöä tai vaihtoehtoisia tapoja viestintä, kuten kaiuttimet, puheen tuottavat laitteet ja/tai aakkostaulut, viestintä kyllä/ei-signaalien kautta.
Ravitsemus
Potilaat ja heidän hoitajansa voivat vastaanottaa tarvittavat tiedot ravitsemusasiantuntijoilta, kuinka suunnitella ruokavaliosi ja syödä pieniä aterioita koko päivän ajan, joista saa riittävästi kaloreita, kuitua ja nestettä, sekä tietoa siitä, mitä ruokia tulisi välttää nielemisvaikeuksien välttämiseksi. Potilaat voivat käyttää imulaitteita ylimääräisen nesteen ja syljen poistamiseen, mikä estää tukehtumisen. Terapeutit voivat auttaa antamaan suosituksia itseruokintaa varten. Puhevammaasiantuntijat voivat auttaa sinua valitsemaan kykyihisi parhaiten sopivat tuotteet. Kun potilas ei voi enää vastaanottaa ravinteita ruoasta, lääkärit voivat suositella syöttöletkun käyttöä. Ruokintaletkun käyttö ehkäisee myös tukehtumis- ja keuhkokuumeriskiä, joka voi aiheutua nesteen hengityksestä keuhkoihin. Luuri ei aiheuta mitään kipu ja antaa potilaille mahdollisuuden syödä itse, jos he haluavat. Tutkijat toteavat, että "ALS-potilailla on krooninen energiansaantivaje" ja ruokahalun lasku. Eläimillä ja ihmisillä tehdyt tutkimukset vahvistavat, että potilaiden tulisi kuluttaa mahdollisimman paljon kaloreita eikä koskaan vähennä kalorien saantiaan. Vuodesta 2012 lähtien painonpudotushoidosta ei ole tarkkoja tietoja.
Palliatiivinen hoito
Sosiaalityöntekijät, omaishoitajat ja saattohoitajat auttavat ALS-potilaita, heidän perheitään ja hoitajia selviytymään lääketieteellisistä, henkisistä ja taloudellisista vaikeuksista, erityisesti taudin pitkälle edenneissä vaiheissa. Sosiaalityöntekijät tukevat taloudellisen tuen saamista, valtakirjan ja testamentin kirjoittamista sekä auttavat löytämään tukea perheille ja omaishoitajille. Hoitajat eivät vain tarjoa sairaanhoito, mutta myös opettaa potilaan perheenjäseniä käyttämään hengityssuojaimia oikein, ruokkimaan ja liikuttamaan potilasta siten, että vältetään kipeät iho-ongelmat ja kireys. Hospice-sairaanhoitajat työskentelevät lääkäreiden kanssa tarjotakseen asianmukaista hoitoa ja apua muissa kotona oleskelevien potilaiden elämänlaadun parantamiseen liittyvissä asioissa. Saattotyöntekijät neuvovat myös potilaita ja heidän omaisiaan kaikissa taudin loppuvaiheeseen liittyvissä asioissa.
Epidemiologia
Useimmissa maissa ALS:n ilmaantuvuutta ei tunneta. Euroopassa tautia sairastaa noin 2,2 ihmistä 100 000 ihmistä kohti vuodessa. Yhdysvalloissa diagnosoidaan vuosittain 5 600 ihmistä, kun taas 30 000 amerikkalaisella on jo tauti. Sivusta amyotrofinen skleroosi kaksi 100 000 ihmisestä kuolee vuosittain. ALS otetaan huomioon harvinainen sairaus, mutta yleisin kaikista motorisista neuronisairauksista, joka vaikuttaa ihmisiin riippumatta rodusta ja etnisestä alkuperästä. ALS kehittyy 1-2 ihmisellä 100 000:ta kohden vuosittain. ALS vaikuttaa jopa 30 000 amerikkalaiseen. Taudin on raportoitu sairastavan 1,2–4,0 henkilöä 100 000 valkoihoista kohden, mutta pienempiä määriä on havaittu muissa etnisissä ryhmissä. Filippiiniläisillä on toiseksi suurin taudin esiintyvyys (1,1-2,8 jokaisesta 100 000:sta). On raportoitu useista "ryhmistä", mukaan lukien kolme amerikkalaista jalkapalloilijaa San Francisco 49ersistä, yli 50 jalkapalloilijaa Italiassa, kolme jalkapalloystävää Etelä-Englannissa ja perhetapauksia (aviomies ja vaimo) Etelä-Ranskassa. Useimmat tutkijat väittävät, että ALS johtuu perinnöllisten ja perinnöllisten sairauksien yhdistelmästä ympäristötekijät, vaikka jälkimmäistä ei ole vahvistettu, toisin kuin taudin riski kasvaa iän myötä.
Tarina
Charles Bell kuvasi taudin ensimmäisen kerran vuonna 1824. Yhdysvalloissa tauti tuli tunnetuksi sen jälkeen, kun se iski kuuluisaan baseball-pelaaja Lou Gehrigiin ja sitten vuonna 2014 kampanjan, jota kutsuttiin jääkauhahaasteeksi. Englantilainen tiedemies August Waller kuvasi ulkomuoto hermosäikeitä vuonna 1850. Vuonna 1869 oireiden ja taustalla olevien neurologisten ongelmien välisen yhteyden kuvasi Jean-Martin Charcot, joka esitteli amyotrofisen lateraaliskleroosin käsitteen työssään vuonna 1874. Vuonna 1881 artikkeli käännettiin Englannin kieli ja julkaistu kolmessa Lectures on Diseases -julkaisussa hermosto". ALS tuli tunnetuksi Yhdysvalloissa vuonna 1939, kun tauti iski baseballlegenda Lou Gehrigiin, joka kuoli kaksi vuotta myöhemmin. 1950-luvulla ALS-epidemia esiintyi Guamin chamorro-kansan keskuudessa. Vuoteen 1991 mennessä tutkijat olivat jo yhdistäneet kromosomin 21 perinnölliseen ALS-muotoon (HALS). Vuonna 1993 havaittiin, että kromosomissa 21 olevalla SOD1-geenillä on tärkeä rooli useissa taudin perinnöllisen muodon tapauksissa. Vuonna 1996 Yhdysvaltain terveysministeriö hyväksyi rilutsolin ALS:n hoitoon. Vuonna 1998 El Escorial -kriteeri otettiin käyttöön standardina ALS-potilaiden luokittelussa kliinisissä tutkimuksissa. Seuraavana vuonna julkaistiin ALS-toiminnallisuusasteikko, josta tuli standardi taudin arvioinnissa kliinisissä tutkimuksissa. Vuonna 2011 useiden C9ORF72-toistojen todettiin olevan ALS:n ja frontotemporaalisen dementian pääasiallinen syy.
Etymologia
Termi "amyotrofinen" tulee kreikan sanasta amyotrophia: a- tarkoittaa "ei", myo tarkoittaa "lihasta" ja trophia tarkoittaa "ravintoa"; siten amyotrofia tarkoittaa "lihasten ravinnon puutetta", joka kuvaa tarkasti ominaispiirre sairaus, lihasten surkastuminen. "Lateraalinen" viittaa ihmisen selkäytimen alueeseen, jossa vahingoittuneet hermosolut sijaitsevat. Tämän alueen rappeutuminen johtaa tiivistymiseen, skleroosiin.
Julkinen tuki ja kulttuurinen viittaus
Elokuussa 2014 Internetissä järjestettiin ALS Ice Bucket Challenge -toiminta ALS-ihmisten tukemiseksi. Mielenosoittajan täytyi täyttää vesiämpäri jäällä, sitten nimetä hänet haastaja ja myös kolme haastamaansa henkilöä. Sitten osallistuja kaatoi vesi- ja jääämpärinsä päälleen. Mutta toimintaan oli mahdollista osallistua toisella tavalla. Jäsen voi lahjoittaa vähintään US$10 (tai vastaavan summan valuutassa) ALS-tutkimukseen Isossa-Britanniassa. Se, joka ei halua kastua kylmä vesi on lahjoitettava vähintään 100 dollaria ALS-tutkimukseen. Elokuun 25. päivään mennessä toiminta on kerännyt 79,7 miljoonaa dollaria, kun vuonna 2013 kerättiin vain 2,5 miljoonaa dollaria. Tähän toimintaan osallistui monia julkkiksia. ALS on keskiössä vuoden 2014 elokuvassa You Are Not You, pääosissa Hilary Swank, Emily Rossum ja Josh Duhamel.
Tutkimus
Kliiniset tutkimukset ovat käynnissä ympäri maailmaa; Luettelo Yhdysvaltojen kliinisistä tutkimuksista löytyy osoitteesta ClinicalTrials.gov. Suurin geneettinen tutkimus, nimeltään MinE-projekti, on edelleen käynnissä. Hanke rahoitetaan julkisella varainkeruulla ja siinä on mukana useita maita. Tutkimuksen II vaihe saatiin päätökseen ja vaihe IIb on edelleen käynnissä nimellä "BEEFIT-ALS". Ensimmäisen tutkimuksen tulokset ovat saatavilla täältä.Tämä tutkimus on kansainvälinen, lumekontrolloitu tutkimus, johon osallistui 680 potilasta. Tämä tekee siitä toistaiseksi suurimman tutkimuksen. Otsanetsumabin vasta-aineen II vaiheen tutkimus on parhaillaan käynnissä. Tämä on suuri tutkimus, jota sponsoroi brittiläinen GlaxoSmithKline. Hadaas-sairaala Israelissa Meneillään vaiheeseen II Kliininen tutkimus BrainStorm Cell Therapeuticsin suorittaman työn tavoitteena on stabiloida ALS-toiminnallisen asteikon parametrit. Kantasolut poistetaan testin aikana luuydintä ihmisen ja erilaistuvat solun vapaaseen tilaan, mikä aktivoi neurotrooppisia tekijöitä. Solut injektoidaan takaisin samaan potilaaseen intratekaalisilla ja lihaksensisäisillä injektioilla. On suunniteltu, että vaiheen II toinen osa suoritetaan useissa Yhdysvaltain laitoksissa, mukaan lukien Mayo Clinic.
Amyotrofista lateraaliskleroosia (ALS) esiintyy kolmella sadasta tuhannesta ihmisestä. Huolimatta lääketieteen nykypäivän edistymisestä tämän patologian kuolleisuus on 100%. On kuitenkin tapauksia, joissa potilaat eivät kuolleet ajan myötä, heidän tilansa vakiintui. Hyvä esimerkki on kuuluisa kitaristi Jason Becker. Hän on taistellut aktiivisesti tätä tautia vastaan yli 20 vuoden ajan.
Mikä on BAS?
Tämän taudin yhteydessä selkäytimen ja yksittäisten aivoosien motoriset neuronit, jotka ovat vastuussa vapaaehtoisista liikkeistä, kuolevat johdonmukaisesti. Ajan myötä tämän diagnoosin saaneiden ihmisten lihakset surkastuvat, koska he ovat jatkuvasti passiivisia. Sairaus ilmenee raajojen, kehon lihasten ja kasvojen halvauksena.
Amyotrofista lateraaliskleroosia kutsutaan amyotrofiseksi lateraaliskleroosiksi, koska neuronit, jotka johtavat impulsseja kaikkiin lihaksiin, sijaitsevat molemmilla puolilla selkäydintä. Taudin viimeinen vaihe diagnosoidaan, kun patologinen prosessi saavuttaa hengitysteitä. Kuolema johtuu lihasten surkastumisesta tai infektiosta. Joissakin tapauksissa hengityslihakset vaikuttavat ennen raajoja. Ihminen kuolee hyvin nopeasti kokematta kaikkia elämän vaikeuksia halvaantuneena.
Monissa Euroopan maissa amyotrofinen lateraaliskleroosi tunnetaan nimellä Lou Gehrigin tauti. Tämä kuuluisa amerikkalainen baseball-pelaaja diagnosoitiin vuonna 1939. Vain parissa vuodessa hän menetti täysin kehonsa hallinnan, hänen lihaksensa olivat uupuneet ja urheilija itse vammautui. Lou Gehrig kuoli vuonna 1941.
Riskitekijät
Vuonna 1865 Charcot (ranskalainen neurologi) kuvasi ensimmäisen kerran tämän taudin. Nykyään vain viisi ihmistä sadasta tuhannesta kärsii siitä kaikkialla maailmassa. Tämän diagnoosin saaneiden potilaiden ikä vaihtelee 20-80 vuoden välillä. Vahvemman sukupuolen edustajat ovat alttiimpia tälle taudille.
10 prosentissa tapauksista ALS-tauti on perinnöllinen. Tutkijat ovat tunnistaneet noin 15 geeniä, joissa on mutaatio vaihtelevassa määrin ilmenee ihmisillä, joilla on tämä patologia. 
Loput 90 % tapauksista ovat sporadisia eli eivät liity perinnöllisyyteen. Asiantuntijat eivät osaa nimetä erityisistä syistä mikä johtaa taudin kehittymiseen. Oletetaan, että jotkin tekijät voivat edelleen lisätä taudin riskiä, nimittäin:
- Tupakointi.
- Työskentele vaarallisella alalla.
- Palvelu armeijassa (tutkijoiden on vaikea selittää tätä ilmiötä).
- Syö torjunta-aineilla kasvatettuja ruokia.
Taudin tärkeimmät syyt
Vakava patologinen prosessi voidaan aloittaa kokonaan erilaisia tekijöitä joita kohtaamme päivittäin oikea elämä. Miksi ALS esiintyy? Syyt voivat olla seuraavat:
- Kehon myrkytys raskasmetalleilla.
- Tarttuvat taudit.
- Tiettyjen vitamiinien puute.
- Sähkövamma.
- Raskaus.
- Pahanlaatuiset kasvaimet.
- Kirurgiset toimenpiteet (vatsan osan poistaminen).
Sairauden muodot
Kohdunkaulan rintakehälle on ominaista patologisen prosessin leviäminen lapaluiden alueelle, käsivarsiin ja kaikkeen olkavyö. Vähitellen ihmisen on vaikea suorittaa tavanomaisia liikkeitä (esimerkiksi nappeja), joihin hänen ei tarvinnut keskittyä ennen sairautta. Kun kädet lakkaavat "tottelemasta", tapahtuu täydellinen lihasatrofia. 
Lumbosakraaliselle muodolle on ominaista tappio alaraajoissa kuin kädet. Vähitellen tämän alueen lihasten heikkous kehittyy, ilmaantuu nykimistä ja kouristuksia. Potilaat alkavat kokea kävelyvaikeuksia, jatkuvasti kompastuvia.
Bulbar muoto on yksi taudin vakavimmista ilmenemismuodoista. Potilaat onnistuvat hyvin harvoin selviytymään yli neljä vuotta ensioireiden alkamisesta. ALS-taudin merkit alkavat puheongelmista ja hallitsemattomista ilmeistä. Potilailla on nielemisvaikeuksia, mikä johtaa täydelliseen kyvyttömyyteen syödä itsenäisesti. Patologinen prosessi, joka kaappaa koko ihmiskehon, vaikuttaa negatiivisesti hengitys- ja hengityselinten toimintaan sydän- ja verisuonijärjestelmät. Tästä syystä tätä muotoa sairastavat potilaat kuolevat ennen halvauksen kehittymistä.
Aivomuodolle on ominaista samanaikainen osallistuminen sekä ylä- että alaraajojen patologiseen prosessiin. Lisäksi potilaat voivat itkeä tai nauraa ilman syytä. Vakavuuden mukaan aivomuoto ei huonompi kuin bulbar, joten kuolema siitä tulee yhtä nopeasti.
Kliininen kuva
Joidenkin tietojen mukaan jopa prekliinisessä vaiheessa noin 80 % motorisista neuroneista kuolee. Vierekkäiset solut ottavat haltuunsa kaiken heidän työnsä. Ne lisäävät vähitellen päätehaarojen määrää, ja niin kutsuttu ionikoodi alkaa muuttua suureksi määräksi lihaksia. Syntyneen ylikuormituksen vuoksi myös nämä neuronit kuolevat. Näin amyotrofinen lateraaliskleroosi alkaa. Taudin oireet eivät ilmene välittömästi motoristen hermosolujen kuoleman jälkeen.
Voi kestää 5-7 kuukautta ennen kuin henkilö kiinnittää huomiota ulkoisia muutoksia kehostasi. Potilailla on yleensä painon lasku ja lihasheikkous, päivittäisten toimintojen toteuttamisessa on vaikeuksia. On vaikeaa liikkua normaalisti, kantaa esineitä käsissäsi, hengittää, niellä ja puhua. Näkyviin tulee kouristuksia ja nykimistä. Tällaiset oireet ovat tyypillisiä monille sairauksille, mikä vaikeuttaa merkittävästi ALS:n diagnoosia varhaisessa kehitysvaiheessa.
Tämä patologia ei koskaan vaikuta järjestelmiin sisäelimet(munuaiset, maksa, sydän), suoliston motiliteettista vastaavat lihakset.
ALS-tauti on luonteeltaan etenevä, ja ajan myötä se kaappaa yhä useampia kehon alueita. Henkilö menettää vähitellen kyvyn liikkua ilman vaikeuksia rikkomuksen vuoksi nielemisrefleksit ruokaa on jatkuvasti Airways mikä aiheuttaa hengityskatkoksia. Viimeisissä vaiheissa elintärkeää toimintaa tuetaan vain keinotekoisen ravinnon ja hengityslaitteen ansiosta. 
Diagnostiikka
Taudin diagnosointi on erittäin vaikeaa. Koko pointti on se alkuvaiheet ALS-taudilla on samanlaisia piirteitä kuin muilla neurologisilla häiriöillä. Vasta potilaan perusteellisen tutkimuksen jälkeen lääkäri voi tehdä lopullisen diagnoosin.
Diagnoosi edellyttää potilaan terveyden monipuolista tutkimusta alkaen anamneesin keräämisestä ja päättyen molekyyligeneettiseen analyysiin. Sitä paitsi sisään ilman epäonnistumista neurologinen tutkimus, MRI, serologinen ja biokemialliset analyysit verta. 
Millainen hoito pitäisi olla?
Tällä hetkellä asiantuntijat eivät pysty tarjoamaan tehokkaita menetelmiä hoitoon. Kaikki lääkäreiden antama apu rajoitetaan helpottamaan taudin ilmenemismuotoja niin paljon kuin mahdollista.
Amyotrofisen lateraaliskleroosin hoitoon kuuluu antikoliiniesteraasilääkkeiden (Galantamiini, Prozerin) ottaminen puheen ja nielemisen laadun parantamiseksi, lihasrelaksantteja (Diazepami), masennuslääkkeitä ja rauhoittavia lääkkeitä. Infektiovaurioissa määrätään antibioottihoito. Kun kova kipu lääkärit suosittelevat ei-steroidisten tulehduskipulääkkeiden ottamista, ne korvataan myöhemmin huumausaineilla.
ainoa tehokas lääke määrätietoinen toiminta on Rilutek. Se ei vain pidennä potilaan elämää, vaan mahdollistaa myös ventilaattoriin siirtymisen viiveen lisäämisen. 
Hyvä hoito parantaa merkittävästi elämänlaatua
Tietenkin jokainen henkilö, jolla on diagnosoitu ALS, tarvitsee asianmukainen hoito. Potilas tutkii kriittisesti tilaansa, koska joka päivä hänen ruumiinsa kirjaimellisesti haalistuu. Lopulta tällaiset ihmiset lakkaavat palvelemasta itseään, kommunikoivat sukulaisten ja ystävien kanssa ja masentuvat.
Poikkeuksetta kaikki ALS-potilaat tarvitsevat:
- Toimivassa sängyssä erityisellä nostomekanismilla.
- WC-istuimessa.
- Pyörätuolissa automaattiohjauksella.
- Viestintävälineissä, esimerkiksi kannettavassa tietokoneessa.
Myös potilaiden ravitsemus vaatii erityistä huomiota. On parempi antaa hyvin nieltyä ruokaa, joka sisältää runsaasti vitamiineja ja proteiineja. Myöhemmin ravitsemus ilman erityistä anturin apua ei ole mahdollista.
Amyotrofinen lateraaliskleroosi kehittyy joillakin ihmisillä nopeasti. Sukulaisten ja ystävien on erittäin vaikeaa, koska henkilö kirjaimellisesti katoaa silmiemme edessä. Usein sairaita hoitavat ihmiset tarvitsevat lisäapua psykologilta sekä rauhoittavien lääkkeiden ottamista.
Ennuste
Jos lääkäri vahvisti amyotrofisen lateraaliskleroosin, oireet vain lisääntyvät päivä päivältä, pahenevat yleinen tila potilas, ennuste tässä tapauksessa on pettymys. Koko modernin lääketieteen historian aikana on kirjattu vain kaksi tapausta, joissa potilaat selvisivät hengissä. Olemme jo puhuneet ensimmäisestä tässä artikkelissa. Ja toinen on kuuluisa fyysikko Stephen Hawking, joka on jatkanut menestyksekkäästi olemassaoloaan tällaisen taudin kanssa viimeiset 50 vuotta elämästään. Tiedemies työskentelee aktiivisesti ja nauttii jokaisesta uudesta päivästä, vaikka hän liikkuukin erityisesti varustetussa tuolissa ja kommunikoi muiden kanssa tietokoneen puhesyntetisaattorin kautta. 
Ennaltaehkäisevät toimenpiteet
NOIN ensisijainen ehkäisy patologiasta ei tarvitse puhua, koska sen esiintymisen tarkat syyt eivät ole vielä selvillä. Sekundaariprevention tulee pyrkiä hidastamaan taudin etenemistä ja pidentää potilaan elinikää. Se sisältää:
- Säännöllinen neurologin konsultaatio ja lääkkeiden käyttö.
- Kaiken täydellinen hylkääminen huonoja tapoja koska ne vain pahentavat ALS-tautia.
- Hoidon tulee olla riittävää ja asiantuntevaa.
- Tasapainoinen ja järkevä ravinto.
Johtopäätös
Tässä artikkelissa puhuimme siitä, mikä on ALS-sairaus. Tämän oireet ja hoito patologinen tila ei pidä jättää huomiotta. Valitettavasti nykyaikainen lääketiede ei voi tarjota tehokasta hoitoa tätä sairautta vastaan. Sukulaisten ja ystävien tulee kuitenkin tehdä kaikkensa parantaakseen jokapäiväinen elämä henkilö, jolla on tämä diagnoosi.
Ranskalainen psykiatri Martin Charcot kuvasi ensimmäisen kerran amyotrofisen lateraaliskleroosin (ALS, "Hehrigin tauti", "motorinen neuronitauti") vuonna 1869.
Yhdysvalloissa ja Kanadassa on toinen termi - "Lou Gehrigin tauti", kuuluisa baseball-pelaaja, joka joutui lopettamaan uransa 36-vuotiaana amyotrofisen lateraaliskleroosin vuoksi.
Mikä on amyotrofinen lateraaliskleroosi?
Amyotrofinen lateraaliskleroosi on hermoston sairaus, joka vaikuttaa nopeasti selkäytimen, aivorungon ja aivokuoren motorisiin hermosoluihin.
SISÄÄN patologinen prosessi osallistua motoriset hermot kallon neuronit (kasvojen, trinaariset, glossofaryngeaaliset).
Amyotrofinen lateraaliskleroosi on harvinainen (2-3 henkilöä 100 000:ta kohti) ja etenee nopeasti.
Lääketieteessä on toinen käsite - amyotrofisen lateraaliskleroosin oireyhtymä. Sen aiheuttaa toinen sairaus, joten hoidon tarkoituksena on tässä tapauksessa poistaa taustalla oleva patologia. Jos potilaalla on ALS-oireita, mutta niiden syitä ei tiedetä, lääkärit eivät puhu oireyhtymästä vaan sairaudesta.
ALS:ssä motoriset neuronit tuhoutuvat, ne lakkaavat lähettämästä signaaleja aivoista lihaksiin, minkä seurauksena jälkimmäiset alkavat heiketä ja surkastua.
Syyt

Syitä, jotka provosoivat tämän taudin ilmaantumista, ei ole vielä vahvistettu. Tiedemiehet tarjoavat kuitenkin useita teorioita:
perinnöllinen
On todettu, että 10-15 prosentissa tapauksista sairaus on perinnöllinen.
Viraalinen
Tämä teoria tuli laajalle levinneeksi 1900-luvun 60-luvulla Yhdysvalloissa ja Neuvostoliitossa. Tällä hetkellä kokeita suoritettiin apinoilla. Eläimiin ruiskutettiin sairaiden ihmisten selkäytimen uutteita. Lisäksi oletettiin, että poliovirus voi aiheuttaa taudin.
Gennaja
Geenihäiriöitä havaitaan 20 prosentilla ALS-potilaista. Ne koodaavat superoksididismutaasi-1-entsyymiä, joka muuttuu vaaralliseksi hermosolut superoksidi hapeksi.
autoimmuuni
Tutkijat suorittivat tutkimusta ja löysivät vasta-aineita, jotka tappavat motorisia hermosoluja. On osoitettu, että näitä vasta-aineita voidaan muodostaa, kun vakavia sairauksia(Hodgkinin lymfooma, keuhkosyöpä jne.).
hermostoa
Tämän teorian ovat kehittäneet brittiläiset tutkijat, jotka uskovat, että ALS:n muodostuminen voi provosoida glia-soluja, jotka ovat vastuussa hermosolujen elintärkeästä toiminnasta. Jos hermopäätteistä glutamaattia poistavien astrosyyttien toiminta heikkenee, Charcotin taudin riski kasvaa useita kertoja.
Riskitekijöistä lääkärit erottavat: perinnöllisen alttiuden, yli 50 vuoden iän, tupakoinnin, lyijyn käyttöön liittyvän työn ja asepalveluksen.
ALS-oireet
Sairauden muodosta riippumatta kaikki potilaat tuntevat lihasheikkoutta, lihasnykitystä ja lihasmassa pienenee.
Lihasheikkous lisääntyy nopeasti, mutta silmän lihakset ja sulkijalihakset Virtsarakko eivät vaikuta.
Taudin varhaisessa vaiheessa esiintyy:
- lihasheikkous nilkoissa ja jaloissa;
- käsien surkastuminen;
- heikentynyt motoriikka ja puhe;
- nielemisvaikeudet;
- lihasten nykiminen;
- kielen, käsivarsien ja hartioiden kouristukset.
ALS:n kehittyessä ilmaantuu nauru- ja itkukohtauksia, tasapaino häiriintyy, kielen surkastuminen ilmenee.
Kognitiiviset toiminnot huononevat vain 1-2 %:lla sairaudesta, muilla potilailla henkinen toiminta ei muutu.
Myöhemmissä vaiheissa potilaalle kehittyy masennus, alkaa hengityskatkoksia ja kyky liikkua itsenäisesti menetetään.
ALS-potilaat lakkaavat olemasta kiinnostuneita läheisistään ja ulkomaailmasta, heistä tulee oikeita, hillitsemättömiä, emotionaalisesti labiileja ja aggressiivisia. Kun hengityslihakset lakkaavat toimimasta, henkilö tarvitsee koneellista ilmanvaihtoa.
Taudin kulku
Aluksi ilmaantuu oireita, jotka estävät henkilöä viettämästä täyttä elämää: lihasten puutuminen, kouristukset, nykiminen, puhevaikeudet. Mutta yleensä alussa on vaikea määrittää näiden häiriöiden tarkkaa syytä.
Useimmissa tapauksissa ALS asetetaan lihasatrofian vaiheeseen.
Vähitellen lihasheikkous leviää ja kattaa uusia kehon osia, potilas ei voi liikkua itsenäisesti, hengitysvaikeudet alkavat.
ALS-potilaat kärsivät harvoin dementiasta, mutta heidän tilansa johtaa vakavaan masennukseen odottaessaan kuolemaa. Viimeisessä vaiheessa ihminen ei voi enää syödä, kävellä ja hengittää yksinään, hän tarvitsee erityisiä lääkinnällisiä laitteita.
Sairauden muodot
Taudin muodot erottuvat vaurioituneiden lihasten sijainnista.
Bulbarnaya
Aivohermot kärsivät (9,10,12 paria).
Potilailla, joilla on ALS-bulbaarimuoto, alkaa olla puheongelmia, he valittavat ääntämisvaikeuksista, heidän on vaikea liikuttaa kieltään.
Taudin edetessä nieleminen häiriintyy, ruoka voi valua ulos nenän kautta. Sairauden myöhäisessä vaiheessa kasvojen ja kaulan lihakset surkastuvat kokonaan, ilmeet katoavat, eivätkä ALS-potilaat voi avata suutaan ja pureskella ruokaa.
kohdunkaulan rintakehä

Sairaus etenee yläraajoissa molemmin puolin.
Aluksi käsissä ilmenee epämukavuutta, ihmisen on vaikea suorittaa monimutkaisia liikkeitä käsillään, kirjoittaa, soittaa soittimia. Tutkimuksessa lääkäri huomaa, että potilaan käsivarsien lihakset ovat jännittyneet ja jännerefleksit lisääntyvät.
Taudin pitkälle edenneissä vaiheissa lihasheikkous etenee ja leviää käsivarsiin ja hartioihin.
lumbosacral
![]()
Ensimmäinen oire on alaraajojen heikkous.
Potilaan on vaikeampaa tehdä työtä seistessä, kiivetä portaissa, ajaa polkupyörää ja kävellä pitkiä matkoja.
Ajan myötä jalka alkaa roikkua, kävely muuttuu, sitten jalkojen lihakset surkastuvat kokonaan, henkilö ei pysty kävelemään, kehittyy virtsan- ja ulosteenpidätyskyvyttömyyttä.
Lähes 50 % potilaista kärsii ALS:n kohdunkaulan ja rintakehän muodosta, joista 25 % on lumbosacral ja bulbar.
Diagnostiikka
Neurologi käyttää pääasiallisina diagnoosimenetelminä:
Aivojen ja selkäytimen MRI
Tällä menetelmällä on mahdollista havaita pyramidirakenteiden rappeutuminen ja aivojen motoristen osien surkastuminen.
Neurofysiologiset tutkimukset
ALS:n havaitsemiseen käytetään TKMS:ää (transkraniaalista magneettista stimulaatiota), ENG:tä (elektroneurografiaa), EMG:tä (elektromyografiaa).
Aivo-selkäydinpunktio
Proteiinipitoisuuden taso (normaali tai kohonnut) määritetään.
Biokemialliset verikokeet
Potilailla, joilla on ALS, havaitaan kreatiinifosfokinaasin 5-kertainen nousu tai enemmän, kreatiniinin ja urean kertyminen sekä ASAT- ja ALAT-arvojen nousu.
Molekyyligeneettinen analyysi
Superoksididismutaasi-1:tä koodaavaa geeniä tutkitaan.
Mutta nämä menetelmät eivät riitä diagnoosin tunnistamiseen rinnakkain käytetty erotusdiagnoosi vahvistaa tai sulkea pois sairauksia:
- aivot: dyscirculatory enkefalopatia, kallon takakuopan kasvaimet, monisysteeminen atrofia.
- selkäydin: kasvaimet, syringomyelia, lymfaattinen leukemia, selkärangan amyotrofia jne.
- lihaksia: myosiitti, okulofaryngeaalinen myodystrofia, myotonia Rossolimo-Steiner-Kurshman.
- ääreishermot: multifokaalinen motorinen neuropatia, Parsonage-Turnerin oireyhtymä jne.
- neuromuskulaarinen synapsi: Lambert-Eatonin oireyhtymä, myasthenia gravis.
Hoito
ALS:iin ei ole parannuskeinoa, mutta on mahdollista hidastaa tämän taudin etenemistä, pidentää elinikää ja lievittää ihmisen tilaa.
Tätä varten käytetään monimutkaista hoitoa:
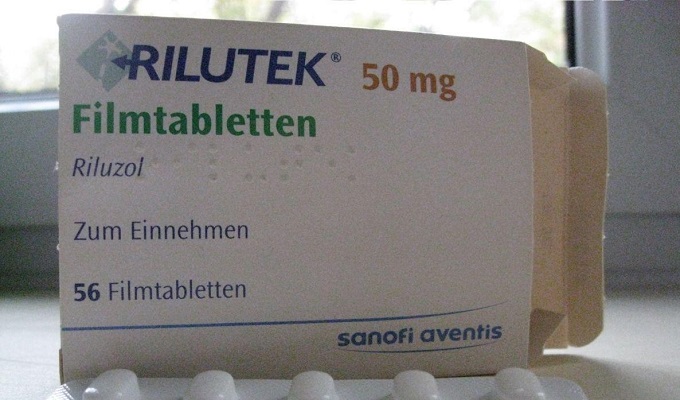
Lääke, jota käytettiin ensimmäisen kerran ALS:n hoidossa Isossa-Britanniassa ja Yhdysvalloissa. Vaikuttavat aineet estävät glutamiinin vapautumisen ja hidastavat hermosolujen vaurioitumista. Lääke on otettava 2 kertaa päivässä 0,05 g.
Lihasrelaksantit ja antibiootit apua lihasheikkouteen. Lihasspasmien ja nykimisen poistamiseksi määrätään Mydocalm, Baclofen, Sirdalud.

Lisäystä varten lihasmassa anabolista "Retabolin" käytetään.
Antibiootit, jos sepsis kehittyy tai tarttuvia komplikaatioita. Lääkärit määräävät fluorokinoloneja, kefalosporiineja, karbopeeneja.
vitamiinit ryhmät B, E, A, C parantamaan impulssia pitkin hermosäikeitä.
Antikoliiniesteraasilääkkeet, jotka hidastavat asetyylikoliinin ("Kalimin", "Prozerin", "Pyridostigmine") tuhoutumisprosessia.

Joissakin tapauksissa sitä käytetään kantasolusiirto. Se estää hermosolujen kuoleman, edistää hermosäikeiden kasvua ja palauttaa hermosolut.
Käytä myöhemmissä vaiheissa masennuslääkkeet ja rauhoittavat lääkkeet, ei-steroidiset kipulääkkeet ja opiaatit.
Jos uni on häiriintynyt, määrätään bentsodiatsepiinivalmisteita.
Liikkumisen helpottamiseksi tuolit ja sängyt erilaisia toimintoja, keppejä, kiinnityskauluksia. Lääkärit neuvovat puheterapiaa. Taudin myöhemmissä vaiheissa tarvitaan syljenpoisto ja sitten trakeostomia, jotta potilas voi hengittää.
Ei-perinteiset ALS-hoitomenetelmät eivät anna positiivista tulosta.
Ennuste ja seuraukset
ALS-potilaiden ennuste haitallisia. Kuolettava lopputulos tapahtuu 2-12 vuoden kuluttua, kuten vaikea keuhkokuume, hengitysvajaus tai muu vakava sairaus Gehrigin taudin aiheuttama.
Sipulimuodossa ja iäkkäillä potilailla ajanjakso lyhenee 3 vuoteen.
Ennaltaehkäisy
Toimenpiteitä ALS-lääkityksen ehkäisemiseksi ei vielä tunneta.
Sairaus etenee erittäin nopeasti, ei ole tapauksia onnistuneesta hoidosta ja kehon motoristen toimintojen palauttamisesta. Lihasheikkous, joka lisääntyy vähitellen, muuttaa täysin ihmisen ja hänen perheensä elämän.
Mutta huolimatta pettymyksestä ennusteesta ja taudin riittämättömästä tutkimuksesta, läheisten tulisi toivoa, että tehokkaita hoitoja kehitetään lähitulevaisuudessa. terapeuttisia menetelmiä hoitoon. Sillä välin on tarpeen ryhtyä toimenpiteisiin ALS-potilaan tilan lievittämiseksi.
amyotrofinen lateraaliskleroosi - vaarallinen sairaus joka voi pysäyttää ihmisen ja johtaa kuolemaan.
Lääkärit eivät vieläkään pysty määrittämään taudin tarkkoja syitä ja löytämään tehokkaita hoitoja. Tällä hetkellä lääke ei voi tehdä muuta kuin lievittää ALS-potilaiden tilaa. Yksikään potilas ei voinut täysin toipua tästä taudista. On tärkeää erottaa "ALS-tauti" "ALS-oireyhtymästä". Toisessa tapauksessa toipumisennuste on paljon parempi.
(ALS), joka tunnetaan myös nimellä Lou Gehrigin tauti, on hitaasti etenevä, parantumaton keskushermoston rappeuttava sairaus. Assosiaatiovaltion mukaan vain puolet Yhdysvaltain asukkaista on kuullut taudista, sama kuvio on havaittavissa muissa maissa.
Yksi tapa saada huomiota amyotrofisen lateraaliskleroosin ongelmaan tuli Ice Bucket Challenge, jossa ihmisten täytyy kastella itseään ämpäri jäävettä ja tehdä lahjoitus. Elokuussa 2014 kampanja saavutti erityisen suosion ympäri maailmaa, sillä se onnistui houkuttelemaan 50 miljoonaa dollaria lahjoituksia ja yli 1,5 miljoonaa osallistujaa. Presidentti ja toimitusjohtaja yritys 3 M Inge Thulin liittyi osallistujamäärään ja kommentoi osallistumistaan toimintaan:
"amyotrofinen lateraaliskleroosi(BAS) - kauhea sairaus. Otin haasteen vastaan 3M:n työntekijämme Allen Wahlgrenin perheeltä, joka on kärsinyt tästä sairaudesta yli 32 vuotta. Hän sai diagnoosin vuoden alussa, ja tänään hän on jo lähes täysin halvaantunut. Tasan vuosi sitten menetimme myös yhden 3M-hammaslääketieteen parhaimmista johtajista, Larry Leerin, joka kuoli ALS:stä. Näin kuinka nopeasti hän "palai loppuun", se oli kauheaa. Ja otin tämän haasteen vastaan en vain Allenin ja Larryn kunniaksi vaan kaikkien perheiden kunniaksi, jotka kohtaavat tämän kauhean taudin."
ALS-taudin syyt
ALS:n syy on joidenkin proteiinien (ubikitiini) mutaatio, jossa esiintyy solunsisäisiä aggregaatteja. Taudin perhemuotoja havaitaan 5 prosentissa tapauksista. Pohjimmiltaan ALS-tauti vaikuttaa yli 46-vuotiaisiin ihmisiin, joista enintään 10% on perinnöllisen muodon kantajia, tutkijat eivät vieläkään pysty selittämään muita tapauksia minkään vaikutuksella. ulkoisista vaikutuksista- ekologia, vammat, sairaudet ja muut tekijät.
Taudin oireet
Taudin varhaisia oireita ovat kouristukset, nykiminen, raajojen tunnottomuus ja heikkous sekä puhevaikeudet, mutta tällaiset merkit koskevat monia sairauksia. Tämä tekee diagnoosista erittäin vaikeaa viimeiseen vaiheeseen asti, sairaus on jo siirtymässä lihasatrofian vaiheeseen.
Alkuperäisiä ALS-leesioita voi esiintyä erilaisia osia elimistöön, kun taas jopa 75 %:lla potilaista sairaus alkaa raajoista, pääasiassa alaraajoista.
Mikä se on? Miten se ilmenee
Taudin ensimmäiset oireet:
.käsien distaalisten osien heikkous, hankaluus suoritettaessa hienoja liikkeitä sormilla, painon lasku käsissä ja nykiminen (lihasnykitys)
.harvemmin sairaus alkaa proksimaalisten käsivarsien ja olkavyön heikkoudella, jalkojen lihasten surkastumalla yhdessä alemman spastisen parapareesin kanssa
On myös mahdollista, että sairaus alkaa bulbar-häiriöillä - dysartria ja dysfagia (25% tapauksista)
Usein yleistyviä kouristuksia (kivuliaita supistuksia, lihaskouristuksia) esiintyy lähes kaikilla ALS-potilailla, ja ne ovat usein ensimmäinen merkki sairaudesta.
ominaisuus kliiniset ilmentymät BASSO
Amyotrofiselle lateraaliskleroosille on ominaista alemman motorisen hermosolun (perifeerinen) ja ylemmän motorisen neuronin (pytamidireitit ja/tai aivojen motorisen aivokuoren pyramidaalisolut) vaurio.
Merkkejä alemman motorisen neuronin vauriosta:
- lihasheikkous (pareesi)
- hyporefleksia (refleksien väheneminen)
- lihasatrofia
- fascikulaatiot (spontaanit, nopeat, ei-rytmiset lihassäikimppujen supistukset)
Merkkejä ylemmän motorisen neuronin vauriosta:
- lihasheikkous (pareesi).
- spastisuus (lisääntynyt lihasjännitys)
- hyperrefleksia (refleksien lisääntyminen)
- patologiset jalka- ja käsimerkit
ALS:lle useimmissa tapauksissa oireiden epäsymmetria.
Atrofoituneissa tai jopa ulospäin koskemattomissa lihaksissa, fascikulaatioita(lihasnykitykset), jotka voivat kuulua paikalliseen lihasryhmään tai olla laajalle levinneitä.
SISÄÄN tyypillinen tapaus taudin puhkeaminen ja lihasten laihtuminen toinen käsi, jossa on kehittynyt adduktion heikkous (adduktio) ja peukalon oppositio (yleensä epäsymmetrisesti), mikä vaikeuttaa peukalolla ja peukalolla tarttumista. etusormet ja johtaa hienomotorisen hallinnan häiriöihin käden lihaksissa. Potilas kokee vaikeuksia poimiessaan pieniä esineitä, kiinnittäessään nappeja, kirjoittaessaan.
Sitten kun tauti etenee, kyynärvarren lihakset ovat mukana prosessissa, ja käsi saa "kynsilevän tassun" ilmeen. Muutamaa kuukautta myöhemmin toisessa kädessä kehittyy samanlainen vaurio. Atrofia, joka leviää vähitellen, kaappaa olkapään ja olkahihnan lihakset.
Samaan aikaan tai myöhemmin sipulilihasten vaurioita kehittyy usein: kielen fascikulaatioita ja atrofiaa, pehmeä kitalaen pareesi, kurkunpään ja nielun lihasten surkastuminen, joka ilmenee dysartriana (puhehäiriöt), dysfagiana (nielemishäiriöinä), syljeneritys.
Mimic ja pureskelu lihaksia vaikuttaa yleensä myöhemmin kuin muihin lihasryhmiin. Sairauden kehittyessä on mahdotonta työntää kieltä, turvottaa poskia ja venyttää huulet putkeen.
Joskus kehittyy pään ojentajien heikkoutta jonka vuoksi potilas ei voi pitää päätään suorana.
Kun mukana kalvon prosessissa havaitaan paradoksaalista hengitystä (hengittäessä vatsa uppoaa, uloshengittäessä se työntyy esiin).
Jalat surkastuvat yleensä ensin etu- ja sivulihasryhmät, mikä ilmenee "riippuvassa jalassa" ja steppage-tyyppisessä kävelyssä (potilas nostaa jalkansa korkealle ja heittää sen eteenpäin laskeen sitä jyrkästi).
Tyypillisesti lihasatrofia on valikoivaa.
- Atrofiaa havaitaan käsissä:
tenara
hypotenaari
luuston väliset lihakset
hartialihakset
- Jaloissa mukana ovat lihakset, jotka suorittavat jalan dorsifleksion.
- Sipulilihaksissa vaikuttaa kielen ja pehmeän kitalaen lihaksiin.
pyramidaalinen oireyhtymä kehittyy pääsääntöisesti ALS:n varhaisessa vaiheessa ja ilmenee jännerefleksien elpymisenä. Tämän jälkeen kehittyy usein matalampi spastinen parapareesi. Käsissä refleksien lisääntyminen yhdistyy lihasten surkastumiseen, ts. ALS:lle on ominaista keskus- (pyramidaalisten) reittien ja perifeerisen motorisen neuronin yhdistetty, samanaikainen vaurio. Pinnalliset vatsan refleksit häviävät prosessin edetessä. Babinskyn oiretta (pohjan katkonainen ärsytys, isovarvas taittuu, muut sormet viuhkamaiset hajaantuu ja taipuvat) havaitaan puolessa sairaustapauksista.
Voi olla aistihäiriöitä. 10 %:lla potilaista havaitaan parestesioita käsivarsien ja jalkojen distaalisissa osissa. Kipu, joskus voimakas, yleensä yöllinen, voi liittyä nivelten jäykkyyteen, pitkittyneeseen liikkumattomuuteen, korkeasta spastisuudesta johtuviin kouristuksiin, kramppeihin (kivuliaisiin lihaskouristeisiin), masennukseen. Herkkyyden menetys ei ole tyypillistä.
Silmän motoriset häiriöt eivät ole tyypillisiä ja esiintyvät taudin loppuvaiheessa.
Lantion elinten toimintahäiriö ei ole tyypillistä, mutta pitkälle edenneissä vaiheissa voi esiintyä virtsan pidättymistä tai inkontinenssia.
Keskivaikea kognitiivinen häiriö(muistin ja henkisen suorituskyvyn heikkeneminen) ilmenee puolella potilaista. 5 %:lla potilaista kehittyy frontaalinen tyyppi, joka voidaan yhdistää parkinsonismiin.
ALS:n ominaisuus on vuoteiden puuttuminen jopa halvaantuneilla vuodepotilailla.
Missä tahansa ne näkyvätkin ALS:n ensimmäiset merkit, lihasheikkous siirtyy vähitellen suurempiin kehon osiin, vaikka ALS:n bulbaarimuodossa potilaat eivät ehkä selviä raajojen täydellisestä pareesista hengityspysähdyksen vuoksi.
Ajan myötä potilas menettää kyvyn liikkua itsenäisesti. ALS-sairaus ei vaikuta henkiseen kehitykseen, mutta useimmiten se alkaa syvä masennus Ihminen odottaa kuolemaa. Sairauden loppuvaiheessa kärsivät myös hengitystoimintoa suorittavat lihakset ja potilaiden elämää on tuettava keinotekoisella keuhkojen ja keuhkojen tuuletuksella. keinotekoinen ravitsemus. ALS:n ensimmäisten merkkien havaitsemisesta kuolemaan kuluu 3–5 vuotta. Laajalti tunnetaan kuitenkin tapauksia, joissa yksiselitteisesti tunnistetun ALS-taudin sairastavien potilaiden tila tasaantui ajan myötä.
KELLÄ ON ALS?
Maailmassa on yli 350 000 ALS-potilasta.
ALS diagnosoidaan 5-7 ihmisellä vuodessa 100 000 asukasta kohden. yli 5 600 amerikkalaisella diagnosoidaan ALS joka vuosi. Se on 15 uutta Bass-tapausta päivässä
ALS voi vaikuttaa keneen tahansa. Ilmaantuvuus (uusien määrä) ALS - 100 000 ihmistä vuodessa
Alle 10 % ALS-tapauksista on perinnöllisiä. ALS voi vaikuttaa sekä miehiin että naisiin ALS vaikuttaa kaikkiin etnisiin ja sosioekonomisiin ryhmiin
ALS voi vaikuttaa nuoriin tai hyvin vanhoihin aikuisiin, mutta se diagnosoidaan useimmiten keski- ja myöhään aikuisiässä.
ALS-potilaat tarvitsevat kalliita laitteita, hoitoa ja jatkuvaa 24/7 hoitoa
90 % hoitotaakasta on ALS-potilaiden perheenjäsenten harteilla. ALS johtaa lopulta fyysisen, emotionaalisen ja taloudelliset resurssit Venäjällä on yli 8 500 ALS-potilasta ja Moskovassa yli 600 ALS-potilasta, vaikka tätä määrää virallisesti jatkuvasti aliarvioidaan. Tunnetuimmat venäläiset, joilla on ALS, ovat Dmitri Šostakovitš ja Vladimir Migulja.
Taudin syitä ei tunneta. ALS:iin ei ole parannuskeinoa. Taudin eteneminen hidastui. Elämän pidentäminen on mahdollista kodin tuulettimen avulla.
Oireet, joita ei voida kliinisesti erottaa klassisesta ALS:sta, voivat johtua seuraavista syistä:
Rakenteelliset vauriot:
parasagittaaliset kasvaimet
foramen magnum kasvaimet
spondyloosi kohdunkaulan selkärangan
Arnold-Chiarin oireyhtymä
hydromyelia
selkäytimen arteriovenoosi anomalia
Infektiot:
bakteeri - tetanus, Lymen tauti
virus - poliomyeliitti, vyöruusu
retrovirusmyelopatia
Myrkytykset, fyysiset tekijät:
myrkyt - lyijy, alumiini, muut metallit.
lääkkeet - strykniini, fenytoiini
sähköisku
röntgenkuvat
Immunologiset mekanismit:
plasmosyyttien dyskrasia
autoimmuuninen polyradikuloneuropatia
Paraneoplastiset prosessit:
parakarsinoomatoosi
paralymfomatoottinen
Aineenvaihduntahäiriöt:
hypoglykemia
hyperparatyreoosi
tyrotoksikoosi
folaatin puutos,
vitamiinit B12, E
imeytymishäiriö
Perinnölliset biokemialliset häiriöt:
androgeenireseptorin vika - Kennedyn tauti
heksosaminidaasin puutos
a-glukosidaasin puutos - Pompen tauti
hyperlipidemia
hyperglysinuria
metyylikrotonyyliglysinuria
Kaikki nämä sairaudet voivat aiheuttaa ALS:n oireita, ja ne tulee ottaa huomioon erotusdiagnoosissa.
Tautiin ei ole tehokasta hoitoa. Ainoa lääke, glutamaatin vapautumisen estäjä rilutsoli (Rilutek), viivyttää kuolemaa 2–4 kuukaudella. Sitä on määrätty 50 mg kahdesti päivässä.
ALS-taudin hoito
Hoidon perustana on oireenmukainen hoito:
- Fysioterapia.
Liikunta. Potilaan tulee mahdollisuuksien mukaan ylläpitää liikunta Taudin edetessä tarvitaan pyörätuolia ja muita erikoislaitteita.
.Ruokavalio. Dysfagia aiheuttaa vaaran, että ruoka pääsee hengitysteihin. Joskus tarvitaan ruokaa letkun kautta tai gastrostomiassa.
- Ortopedisten laitteiden käyttö: kohdunkaulan kaulus, erilaiset lastat, laitteet esineiden tarttumiseen.
Kouristuksilla (kivuliasta lihaskouristukset): kiniinisulfaatti 200 mg kahdesti päivässä tai fenytoiini (difeniini) 200-300 mg / vrk tai karbamatsepiini (Finlepsin, Tegretol) 200-400 mg / vrk ja/tai E-vitamiini 400 mg kahdesti päivässä, samoin magnesiumvalmisteina verapamiili (Isoptin).
.Kun spastisuus: baklofeeni (baclosan) 10-80 mg / vrk tai titsanidiini (Sirdalud) 6-24 mg / vrk, samoin kuin klonatsepaami 1-4 mg / vrk tai memantiini 10-60 mg / vrk.
- Syljeneritystä varten atropiinia 0,25-0,75 mg kolme kertaa päivässä tai hyoskiinia (Buscopan) 10 mg kolme kertaa päivässä.
Jos syöminen on mahdotonta nielemishäiriön vuoksi, tehdään gastrostomia tai asetetaan nenämahaletku. Varhainen perkutaaninen endoskooppinen gastrostomia pidentää potilaiden elinikää keskimäärin 6 kuukaudella.
- klo kipuoireyhtymät käyttää koko kipulääkkeiden arsenaalia. Mukaan lukien huumausainekipulääkkeet loppuvaiheessa.
- Joskus tilapäistä paranemista tuovat antikoliiniesteraasilääkkeet (neostigmiinimetyylisulfaatti - proseriini).
Cerebrolysiini suurina annoksina (10-30 ml IV tiputus 10 päivän ajan toistuvina kursseina). On olemassa useita pieniä tutkimuksia, jotka osoittavat serebrolysiinin hermoja suojaavan tehon ALS:ssä.
. Masennuslääkkeet: Sertaliini 50 mg/vrk tai Paxil 20 mg/vrk tai Amitriptyliini 75-150 mg/vrk sivuvaikutukset- se aiheuttaa suun kuivumista, mikä vähentää liiallista syljeneritystä (syljeneritystä), joka usein piinaa ALS-potilaita).
.Hengityshäiriöiden sattuessa: keinotekoinen ilmanvaihto keuhkoja sairaalassa ei pääsääntöisesti suoriteta, mutta jotkut potilaat ostavat kannettavat ventilaattorit ja suorittavat koneellisen ilmanvaihdon kotona.
- Kasvuhormonin eli neurotrofisten tekijöiden käyttöä ALS:ssä kehitetään parhaillaan.
- Viime aikoina kantasoluterapiaa on kehitetty aktiivisesti. Tämä menetelmä lupaa lupaavaa, mutta on vielä tieteellisten kokeiden vaiheessa.
Lääkkeet lääkkeet, jotka auttavat hidastamaan ALS-tautia:
Nykyaikainen lääketiede kehittyy jatkuvasti. Tutkijat luovat uusia lääkkeitä aiemmin parantumattomiin sairauksiin. Nykyään asiantuntijat eivät kuitenkaan pysty tarjoamaan riittävää hoitoa kaikkia vaivoja vastaan. Yksi näistä patologioista on ALS-tauti. Tämän taudin syitä ei ole vielä tutkittu, ja potilaiden määrä kasvaa vain joka vuosi. Tässä artikkelissa tarkastellaan lähemmin tätä patologiaa, sen tärkeimpiä oireita ja hoitomenetelmiä.
yleistä tietoa
Amyotrofinen lateraaliskleroosi (ALS, Charcotin tauti) on vakava hermoston patologia, jossa selkäytimen ja aivokuoren niin sanotut motoriset neuronit ovat vaurioituneet. Tämä on krooninen ja parantumaton sairaus, joka johtaa vähitellen koko hermoston rappeutumiseen. Sairauden viimeisissä vaiheissa ihminen tulee avuttomaksi, mutta samalla hän säilyttää mielen selkeyden ja mielenterveyden.
ALS-sairaus, jonka syitä ja patogeneesiä ei ole täysin tutkittu, ei eroa erityisissä diagnoosi- ja hoitomenetelmissä. Tiedemiehet jatkavat aktiivisesti sen tutkimista tänään. Nyt tiedetään varmasti, että tauti kehittyy pääosin 50-70-vuotiailla, mutta tapauksia on myös aikaisempia vaurioita.
Luokittelu
Taudin ensisijaisten ilmenemismuotojen sijainnista riippuen asiantuntijat erottavat seuraavat muodot:
- Lumbosacral muoto (alaraajojen motorisessa toiminnassa on häiriö).
- Bulbaarimuoto (joihinkin aivojen ytimiin vaikuttaa, mikä edellyttää luonnetta).
- Kohdunkaulan muoto ( ensisijaisia oireita ilmenevät tavanomaisen motorisen toiminnan muutoksilla Yläraajat).
Toisaalta asiantuntijat erottavat kolme muuta ALS-sairauden tyyppiä:
Miksi ALS esiintyy?
Syyt tämä sairaus valitettavasti edelleen huonosti ymmärretty. Tutkijat tunnistavat tällä hetkellä useita tekijöitä, joiden läsnä ollessa sairastumisen todennäköisyys kasvaa useita kertoja:
ALS-sairaus. Oireet
Valokuvia tätä tautia sairastavista potilaista voi katsella erikoistuneissa hakuteoksissa. Kaikilla heillä on vain yksi yhteinen piirre - ulkoisia oireita taudin myöhemmissä vaiheissa.
Mitä tulee ensisijaisiin kliiniset oireet patologioita, ne aiheuttavat hyvin harvoin potilaiden valppautta. Lisäksi potentiaaliset potilaat selittävät ne usein jatkuvalla stressillä tai työrutiinin levon puutteella. Seuraavat ovat taudin alkuvaiheessa ilmeneviä oireita:
- lihas heikkous;
- dysartria (puhumisvaikeudet);
- usein esiintyvät lihaskouristukset;
- raajojen tunnottomuus ja heikkous;
- lieviä lihasnykistyksiä.
Kaikkien näiden merkkien tulisi varoittaa kaikkia ja olla syy ottaa yhteyttä asiantuntijaan. Muuten tauti etenee, mikä lisää useita kertoja komplikaatioiden todennäköisyyttä.
Taudin kulku
Miten ALS kehittyy? Sairaus, jonka oireet on lueteltu yllä, alkaa aluksi lihas heikkous ja raajojen puutuminen. Jos patologia kehittyy jaloista, potilailla voi olla vaikeuksia kävellä, kompastella jatkuvasti.
Jos sairaus ilmenee yläraajojen rikkoutuessa, alkeellisten tehtävien suorittamisessa (paidan nappiminen, avaimen kääntäminen lukossa) on ongelmia. 
Kuinka muuten ALS voidaan tunnistaa? Taudin syyt 25 %:ssa tapauksista ovat vauriossa ydinjatke. Aluksi on vaikeuksia puheessa ja sitten nielemisessä. Kaikki tämä aiheuttaa ongelmia ruoan pureskelun kanssa. Tämän seurauksena henkilö lopettaa normaalin syömisen ja laihtuu. Tämän seurauksena monet potilaat joutuvat siihen masennus, koska sairaus ei yleensä vaikuta
Joillakin potilailla on vaikeuksia muodostaa sanoja ja jopa normaali keskittyminen. Tämän tyyppiset pienet rikkomukset selittyvät useimmiten huonolla hengityksellä yöllä. Lääketieteen työntekijät jo nyt pitäisi kertoa potilaalle taudin kulun piirteistä, hoitovaihtoehdoista, jotta hän voi etukäteen tehdä tietoisen päätöksen tulevaisuudestaan.
Suurin osa potilaista kuolee hengitysvajaus tai keuhkokuume. Yleensä kuolema tapahtuu viiden vuoden kuluessa taudin toteamisesta.
Diagnostiikka
Vain asiantuntija voi vahvistaa tämän taudin esiintymisen. Tässä asiassa ensisijaisena roolina on olemassa olevan asiantunteva tulkinta kliininen kuva tietyssä potilaassa. ALS-taudin erotusdiagnoosi on erittäin tärkeä.
Millainen hoito pitäisi olla?
Valitettavasti lääketiede ei nykyään pysty tarjoamaan tehokasta hoitoa tätä sairautta vastaan. Miten ALS voidaan parantaa? Hoidon tulee ensisijaisesti suunnata patologian etenemisen hidastamiseen. Näihin tarkoituksiin käytetään seuraavia toimenpiteitä:

Kantasoluhoito
Monissa Euroopan maissa ALS-potilaita hoidetaan nyt omilla kantasoluillaan, mikä myös hidastaa taudin etenemistä. Tämän tyyppisen terapian tarkoituksena on parantaa aivojen ensisijaisia toimintoja. siirretty vaurioalueelle, palauttaa hermosoluja, parantaa aivojen hapen saantia ja edistää uusien verisuonten ilmaantumista.
Itse kantasolujen eristäminen ja niiden suora siirto suoritetaan pääsääntöisesti avohoidossa. Hoidon jälkeen potilas pysyy sairaalassa vielä 2 päivää, jossa asiantuntijat seuraavat hänen tilaansa. 
Itse toimenpiteen aikana soluja injektoidaan selkäydinneste ne eivät saa lisääntyä itsestään kehon ulkopuolella, ja uudelleenistutus suoritetaan vasta perusteellisen puhdistuksen jälkeen.
On tärkeää huomata, että tällainen hoito voi merkittävästi hidastaa ALS-tautia. Potilaiden valokuvat 5-6 kuukautta toimenpiteen jälkeen todistavat selvästi tämän väitteen. Se ei auta pääsemään taudista kokonaan eroon. Valitettavasti on myös tapauksia, joissa hoito ei anna lainkaan vaikutusta.
Johtopäätös
Tämän taudin ennuste on lähes aina epäsuotuisa. Edistyminen liikehäiriöt väistämättä (2-6 vuotta).
Tässä artikkelissa puhuimme siitä, mikä on sellainen patologia kuin ALS. Sairautta, jonka oireet eivät välttämättä ilmene pitkään aikaan, on tällä hetkellä mahdotonta parantaa kokonaan. Tutkijat ympäri maailmaa jatkavat aktiivisesti tämän taudin, sen syiden ja kehityksen nopeuden tutkimista yrittäen löytää tehokkaan lääkkeen.

