DEDIČNÉ CHOROBY NERVOVÉHO SYSTÉMU
PREDNÁŠKA 16
Degeneratívne ochorenia s prevládajúcou léziou nervovosvalového aparátu tvoria najväčšiu skupinu všetkých dedičných ochorení.
Mimoriadne dôležité, a často rozhodujúce v diagnostike nervovosvalových ochorení, sú výsledky elektrofyziologických a biochemický výskum. Rovnako veľký je význam patomorfologických nálezov. Štúdium svalovej biopsie pod svetelným mikroskopom pomáha odlíšiť myogénnu a neurogénnu atrofiu. Histochemické vyšetrenie je nevyhnutné na zistenie metabolických svalových lézií, a elektrónová mikroskopia objavil celú veľkú triedu chorôb – neprogresívne myopatie.
Jemné naťahovanie postihnutých kĺbov znižuje výskyt kontraktúr a tým vedie k pomalšej progresii ochorenia. V tomto prípade sa vykonávajú rôzne špecifické cvičenia a tlakový obväz Urias sa osvedčil pri zhoršenej hĺbke končatín a často sa považuje za domácu liečbu pri prepustení. Senzomotorický tréning pacientov so svalovými ochoreniami sa realizuje v individuálnej alebo skupinovej terapii a zameriava sa na koordináciu a jemnú motoriku.
Veľmi dôležitá metóda Liečba ľudí so svalovými chorobami spočíva v obnovení schopnosti opäť viesť auto. Na tento účel funguje spolupráca s externou autoškolou na oddelení pracovnej terapie a jazda v Bad Wildungen sa vykonáva vo vhodnom invalidnom vozidle s vyškoleným autoinštruktorom.
Progresívne svalové dystrofie. Pojem svalové dystrofie je skupina geneticky podmienených porúch charakterizovaných progresívnym degeneratívne zmeny vo svalových vláknach bez primárnej patológie periférneho (dolného) motorického neurónu.
Rôzne formy sa navzájom líšia typmi dedičnosti, načasovaním nástupu procesu, povahou a rýchlosťou jeho priebehu, zvláštnosťou topografie svalovej atrofie, prítomnosťou alebo absenciou pseudohypertrofie a retrakcií šliach atď. znamenia.
Doplnkovým prístupom pri rehabilitácii svalových ochorení je poskytovanie adekvátneho poradenstva. To platí nielen pre ortézy, ale pre všetky ostatné pomôcok doma je mimoriadne užitočná Cielená pomoc zdravotne postihnutým ľuďom so svalovými chorobami, invalidné vozíky sú presne zmerané a prispôsobené potrebám ľudí trpiacich svalovými poruchami. Sprchové kreslo, vaňový výťah, invalidné vozíky, rampy pre invalidné vozíky a ďalšie.
Klinika fyzikálnej terapie využíva rôzne procedúry na uvoľnenie pacienta. Okrem toho môže toto oddelenie liečby znížiť často malé symptómy bolesti u pacientov so svalovými poruchami. Logopédia je ďalším odborom rehabilitácie na liečbu svalových ochorení. Dalsia diagnoza by sa mala urobit aj tu s laryngoskopiou alebo iste röntgenové štúdie. V kvalifikovanej rehabilitačnej ambulancii pre liečbu svalových ochorení je dôležitým predpokladom holistický terapeutický prístup k tomuto ochoreniu.
Väčšina svalových dystrofií je dobre klinicky študovaná Detailný popis vyrobený koncom minulého storočia. Ale napriek takmer storočnej histórii štúdia myodystrofie otázky ich patogenézy a liečby zostávajú dodnes nevyriešené. Veľké nádeje sa vkladajú do molekulárnej genetiky, pomocou ktorej sa už podarilo určiť umiestnenie génov mnohých nozologických foriem.
Patria sem aj absolventi psychológie fakulty. Postihnuté osoby trpia v dôsledku fyzického nedostatku a súvisiacich dôsledkov v súkromí - Stredná dĺžka života sa často spája s depresívnych nálad a psychosomatické ťažkosti. Podpora individuálnych rozhovorov alebo učenie sa o relaxačných technikách ako autogénny tréning alebo progresívna svalová relaxácia sú tu veľmi nápomocné.
Otázky týkajúce sa zákona o ošetrovateľstvo, ťažké zdravotné postihnutie, pracovisko, kontakt so zamestnávateľom pre pracovnú rehabilitáciu či začlenenie do firmy tu treba uviesť ako časté témy sociálneho poradenstva. Často je navyše aj cielené poradenstvo v rámci konzultácií často kladené otázky pre pacientov so svalovými poruchami. Napokon, plán terapie počas rehabilitácie v Bad Wildungen zahŕňa aj koordináciu kvalifikovaného ortopéda. Vicerek je vedúcim lekárom ortopedického oddelenia kliniky v Hombergu v Bad Wildungene.
Diagnóza svalových dystrofií často predstavuje veľké ťažkosti. Existuje veľká variabilita klinických prejavov a malý počet rodinných príslušníkov sťažuje určenie typu dedičnosti.
Charakteristickým motorickým defektom u pacientov so svalovými dystrofiami je „kačacia“ chôdza: pacient chodí kolísavo na jednu stranu. Súvisí najmä so slabosťou gluteálnych svalov, predovšetkým stredných a malých, ktoré fixujú panvu voči stehennej kosti. Následkom ochorenia dochádza k záklonu panvy k nepodporujúcej nohe (Trendelenburgov fenomén) a kompenzačnému záklonu tela v r. opačná strana(Duchenneov fenomén). Pri chôdzi sa strana svahu neustále mení. Tieto zmeny je možné skontrolovať aj v Trendelenburgovom teste tak, že pacienta požiadate, aby zdvihol jednu nohu a pokrčil ju v pravom uhle v kolenných a bedrových kĺboch: panva na strane zdvihnutej nohy klesá (a nedvíha sa ako normálne) v dôsledku slabosti svalu gluteus medius opornej nohy.
Klinika uskutočňuje pravidelné školenia, ako aj účasť na príslušných kongresoch a sympóziách. Toto stretnutie sa uskutočnilo najmä. Často dochádza aj k zvýšeniu svalových enzýmov. Choroby sa zvyčajne vyvíjajú pomaly, ale rýchlo sa vyskytujú choroby. Ak máte podozrenie, že máte myopatiu alebo ste zapojený do známeho neuromuskulárneho ochorenia, môžete sa objednať.
Liečba závisí od konkrétny dôvod, zápalové ochorenie svalov môže, napríklad, Napríklad rôzne imunoterapeutické činidlá. Podrobný prehľad v génových tabuľkách nervovosvalových chorôb možno nájsť rôzne genetické svalové poruchy.
Pacient s ťažkou svalovou slabosťou proximálnych svalov sa zdvihne z vodorovnej polohy a takmer sa neprevráti na brucho, potom, keď si položí ruky na podlahu, postaví sa na všetky štyri a potom si položí ruky na holene, potom na boky , postupne sa narovnáva. Tento jav „vyberať si na vlastnú päsť“ sa nazýva Gowersov manéver. Často je spojená so slabosťou svalov gluteus maximus.
Lieky, ktoré môžu spôsobiť alebo zvýšiť svalové ochorenie
V našej praxi sú dostupné všetky potrebné diagnostické možnosti. Je v tom zapojená prax, organizujú sa pravidelné školenia v oblasti nervovosvalových ochorení. Dotknuté osoby sa môžu zaregistrovať aj do registra pacientov. Registre pacientov sú dôležitým prvým krokom k lepšiemu pochopeniu výskumu vývoja chorôb a liečby.
Poškodenie srdcového svalu
V tomto článku je zhrnuté možné zapojenie srdcového svalu do myopatie a iných neuromuskulárnych ochorení.Duchennova myodystrofia. Pseudohypertrofická Duchennova svalová dystrofia sa vyskytuje častejšie ako všetky ostatné ochorenia svalového systému (30 na 100 000 živonarodených detí). Vyznačuje sa skorým nástupom a malígnym priebehom. Klasický obraz sa prejavuje zmenou chôdze u dieťaťa vo veku 2-5 rokov, vo veku 8-10 rokov už deti chodia s ťažkosťami, do 14-15 rokov sú väčšinou úplne imobilizované. Deti majú viac nízky vek počiatočné príznaky sa prejavujú zaostávaním motorického vývoja: neskôr začínajú chodiť, nevedia behať a skákať. Pacienti zomierajú v 2. alebo 3. dekáde života.
Máme na mysli všetky nervovosvalové ochorenia. Podľa Waltonovej klasifikácie existuje 800 foriem. Tento príznak môže byť obmedzený na niekoľko svalových skupín alebo, v závislosti od choroby, môže byť nájdená celá svalovina. Ubúdanie svalov, redukcia svalová hmota A svalová slabosť sú hlavnými príznakmi nervovosvalových ochorení. Lieky sa označujú ako lieky na zníženie atrofie muskuloskeletálneho svalstva. Zatiaľ však neposkytujú priamu diagnózu. Existuje celý rad dôvodov, ktoré môžu viesť k čiastočnému zníženiu svalovej hmoty, čo čiastočne súvisí s celým telom.
Jedným z prvých príznakov ochorenia je zhutnenie lýtkových svalov a postupné zväčšovanie ich objemu v dôsledku pseudohypertrofie. Atrofia svalov stehna, panvového pletenca je často maskovaná dobre vyvinutým podkožným tukovým tkanivom. Postupne sa proces uberá smerom nahor a šíri sa za ramenný pletenec, chrbtové svaly a potom do proximálnych častí ramien.
Skutočná príčina takejto svalovej atrofie alebo svalovej slabosti môže spočívať vo vzdialených, úplne odlišných oblastiach tela. Príčiny môžu byť v nervových bunkách miecha, zodpovedný za pohyb, v zásobovacích nervoch, keď sa nerv prenáša do svalu, alebo v samotných svaloch.
Liečba neuromuskulárnych ochorení
Existujú sľubné lieky na liečbu myozitídy, myasténie gravis a endokrinnej myopatie. Prvé prístupy k spomaleniu sú možné s laterálna skleróza amyotrofný. V prípade dedičných svalových dystrofií a spinálnych svalových atrofií nebola doteraz stanovená kauzálna liečba. Sú zamerané na špeciálne očakávania do budúcnosti génová terapia. Pri všetkých nervovosvalových ochoreniach je symptomatológia dôsledná fyzioterapeutická podpora v kombinácii s ortopedickými opatreniami, v niektorých prípadoch s podporou dýchania a v niektorých prípadoch s možnosťou transplantácie srdca.
IN terminálne štádium svalová slabosť sa môže rozšíriť na svaly tváre, hltanu, dýchacích svalov.
V pokročilom štádiu ochorenia existujú charakteristické príznaky, Ako " kačacia prechádzka“; zvýraznená bedrová lordóza, pterygoidné lopatky, symptóm „uvoľneného ramenného pletenca“. Typické sú skoré svalové kontraktúry a retrakcie šliach, najmä Achillových šliach. Kolenné reflexy vypadnú skoro a potom reflexy z horných končatín.
Liečba zahŕňa aj kompenzáciu fyzických obmedzení vhodnými prostriedkami. Nemecká spoločnosť pre svalové choroby zriadila nezávislé informačné centrum. Je tu aj možnosť vyskúšať si produkty doma v dvoch štandardných bytoch alebo stráviť len pár dní v bezbariérových bytoch.
Svalové poruchy Nezávislé svalové poruchy sú relatívne zriedkavé. Na druhej strane sa často stáva, že svaly sú postihnuté inými základnými ochoreniami, najmä chorobami nervový systém; infekčné choroby ako je týfus a tuberkulóza; Parazitárne ochorenia, Trinity a ošípané červy, ako aj hormonálne poruchy.
Pseudohypertrofia sa môže vyvinúť nielen v oblasti gastrocnemia, ale aj v gluteálnych, deltových, brušných a jazykových svaloch. Veľmi často srdcový sval trpí typom kardiomyopatie. Poruchy rytmu srdcovej činnosti, rozšírenie hraníc srdca, hluchota tónov, Zmeny EKG. Najviac je akútne srdcové zlyhanie spoločná príčinaúmrtia na Duchennovú myodystrofiu. Pri pitve sa zistí fibróza a tuková infiltrácia srdcového svalu.
Jednoduchá strata svalov sa často vyskytuje v dôsledku zachovania alebo odpočinku, Svalová kontrakcia môže byť spôsobená zrastmi alebo zranením v dôsledku zlého obehu - V dôsledku asociácií dusenia. Príčinou svalových bolestí môže byť svalový reumatizmus, presilenie, prechladnutie, metabolické poruchy alebo presilenie niektorých svalových skupín v dôsledku deformácie kostry.
Myóm - benígny nádor svalové tkanivo. Polymyozitída: Svalové ochorenie s podobnými príznakmi ako dermatomyozitída, ale bez kožná vyrážka. Pokiaľ ide o príčinu, predpokladá sa, že porušenie sa týka autoagresie, ako utrpenia, ktoré prichádza v dôsledku útoku tela na jeho vlastné štruktúry.
Často dochádza k porušeniu motility gastrointestinálneho traktu.
Bežným príznakom je znížená inteligencia. Zaujímavosťou je, že v niektorých rodinách sa oligofrénia prejavuje ostro, v iných relatívne mierne. Zmena vyšších psychických funkcií väčšinou neprogreduje a nekoreluje so závažnosťou svalového defektu. Nedá sa to vysvetliť len pedagogickým zanedbávaním chorých detí, ktoré predčasne odchádzajú z detských kolektívov, nenavštevujú MATERSKÁ ŠKOLA a škola z dôvodu motorického postihnutia. CT a MRI často odhalia cerebrálnu atrofiu, pravdepodobne spojenú s narušeným prenatálnym vývojom mozgu.
Existuje viac ako 200 foriem svalových chorôb, niektoré sú pomenované po ich objaviteľoch a iné podľa poruchy. Najbežnejšie sú tri podskupiny. Progresívna svalová dystrofia Spinálna svalová atrofia Neurónová svalová atrofia. V klinickej praxi sú tieto ochorenia zriedkavé a v rukách neurológa absolútne nevyhnutné.
Genetický defekt v chromozóme 19 je lézia svalové bunky ktoré sa dodnes nedajú liečiť. Dôvodom je pravdepodobne zmena membránového systému svalových buniek. Mystická dystrofia môže viesť k úplnému zničeniu svalových buniek. Najprv sú postihnuté svaly na tvári, rukách, predlaktiach, dolných končatinách a chodidlách. Choroba sa môže vyskytnúť u mužov a žien všetkých vekových skupín.
Často sa u detí vyvinie adiposogenitálny syndróm, niekedy aj iné príznaky endokrinnej insuficiencie. Často nájdete zmeny v kostrovom systéme: deformácia chodidiel, hrudník, chrbtica, difúzna osteoporóza.
Výrazná vlastnosť Duchennova forma je vysoký stupeň hyperenzýmia už zapnutá skoré štádia vývoj procesu. Hladina enzýmu špecifického pre svalové tkanivo - kreatinín fosfokinázy - v krvnom sére teda môže presiahnuť desiatky a dokonca stokrát normálny výkon. Prudké (10-100-násobné) zvýšenie kreatinínfosfokinázy (CPK) v neuromuskulárnej patológii by malo vyvolať diskusiu predovšetkým o nasledujúcich ochoreniach: Duchennova choroba, Beckerova choroba, poliomyozitída a dermatomyozitída, paroxyzmálna myoglobulinúria, distálna myodystrofia. Až v pokročilých štádiách ochorenia sa stupeň hyperenzýmy postupne znižuje. Existujú správy o zvýšení CPK v štádiu vnútromaternicového vývoja.
Typická je oneskorená svalová relaxácia po svalové napätie. Dôsledkom je svalová slabosť a obmedzenie pohybu v nohách, rukách a rukách, zhoršenie jemnej motoriky. Niektoré boľavé svaly sú obzvlášť silné, pretože majú typické zloženie tkaniva svalové vlákna uložené v tukovom a spojivovom tkanive.
Označuje sa tiež ako ovplyvňovanie miechy svalový kŕč. Je ich až 30 rôzne formy spinálna svalová atrofia. Najbežnejšou formou je proximálna spinálna svalová atrofia, ktorá je tu opísaná. Označuje sa nasledovne za začiatkom trupu.
Duchennova svalová dystrofia sa prenáša recesívnym spôsobom viazaným na X. Gén sa nachádza na krátkom ramene X chromozómu. Frekvencia génových mutácií je pomerne vysoká (30%), čo vysvetľuje veľké množstvo sporadické prípady.
Mutácia (najčastejšie delécia) vedie k sexuálnej alebo takmer úplnej absencii génového produktu - štruktúrneho proteínu dystrofika. Fyziologická úloha dystrofia nebola úplne preukázaná. Nachádza sa vo vysokých koncentráciách v sarkoléme, zrejme hrá úlohu pri udržiavaní integrity tejto membrány. Neprítomnosť dystrofie spôsobuje štrukturálne zmeny v sarkoléme, čo následne vedie k strate intracelulárnych zložiek a zvýšenému vstupu vápnika, čo v konečnom dôsledku vedie k smrti myofibríl. Predpokladá sa, že nedostatok dystrofických v synaptických zónach kortikálnych neurónov je príčinou mentálnej retardácie.
Dôvodom je pravdepodobne genetická chyba. Nervové bunky v mieche sú infikované predné skvamózne bunky. Príčinou je pravdepodobne nervový systém nervov. To platí len pre motorický nervový systém. Časti nervového systému, ktoré sú zodpovedné za pocit dotyku, vnímanie bolesti a teploty, zostávajú nedotknuté. Funkcia močového mechúra a konečník sa nezhoršuje.
Označenie tiež predstavuje stratu svalov v dôsledku nervov. Príčinou je takmer vždy genetický defekt, obaly nervových vlákien abnormálne zhrubnú, prípadne sa zničia samotné obaly nervov. Zapnuté nervové vlákna postihuje ruky a nohy.
Pre lekárske genetické poradenstvo je veľmi dôležité vytvorenie heterozygotného nosiča. S Duchennovou myodystrofiou u heterozygotov v približne 70 % prípadov subklinická a niekedy dokonca jasné znaky svalová patológia - určité zhutnenie až zväčšenie lýtkových svalov, rýchla svalová únava pri fyzickej námahe, zmeny EMG a patomorfologické vyšetrenie vzoriek svalovej biopsie. Najčastejšie heterozygotní nosiči vykazujú zvýšenie aktivity kreatinínfosfokinázy.
Intenzita rýchlosti nervového vedenia sa spomaľuje. Počnúc dolnými končatinami so stratou svalov a súčasnou svalovou slabosťou. Symptómy potom stúpajú na dolných končatinách, neskôr postihujú paže a predlaktia. Senzorické poruchy sú nízke. Možné sú autonómne poruchy, ako je príliš veľké alebo príliš malé potenie a narušenie základného prietoku krvi. Môžu sa vyskytnúť menšie spastické príznaky v nohách.
Svalová slabosť je výsledkom svalovej slabosti, kým svaly nie sú plne funkčné, čo vedie k závažným závažným pohybovým obmedzeniam v nohách, rukách a rukách. Svaly sú veľmi dôležitou súčasťou nášho tela. Bez svalov telo stráca schopnosť pohybovať sa a vykonávať rôzne akcie. Vlastne bez človeka svalový systém asi neprežiješ. Je to spôsobené tým, že väčšina orgánov v zažívacie ústrojenstvo pozostávajú zo svalov a dokonca aj samotné srdce, ktoré pumpuje krv, je tiež sval.
Pri klinickom obraze Duchennovej myodystrofie u žien by sa mala najskôr vylúčiť možnosť anomálie na chromozóme X - Shereshevsky-Turnerov syndróm (XO), Morrisov syndróm (XY) alebo mozaikovitosť u týchto syndrómov.
Duchennova svalová dystrofia, ktorá sa začína rozvíjať už v prenatálnom období, je v podstate vrodená myopatia a možno ju diagnostikovať krátko po narodení vykonaním svalovej biopsie a stanovením aktivity CPK.
Myodystrofia Becker. Spolu s ťažkou, malígnou formou X-viazanej Duchennovej myodystrofie existuje aj benígna forma - Beckerova choroba. Z hľadiska klinických príznakov je veľmi podobná Duchennovej forme, ale spravidla začína neskôr - vo veku 10-15 rokov, mierne prúdi, pacienti zostávajú dlhodobo práceneschopní, vo veku 20 rokov -30 rokov a neskôr môžu stále chodiť. Plodnosť nie je znížená, takže choroba sa niekedy vyskytuje vo viacerých generáciách rodiny: chorý muž prenáša chorobu na svojho vnuka prostredníctvom svojej dcéry („dedinský efekt“). Počiatočné príznaky, podobne ako pri Duchennovej chorobe, sa prejavujú slabosťou svalov panvového pletenca, potom proximálnych dolných končatín. Pacienti menia chôdzu, pociťujú ťažkosti pri stúpaní po schodoch, pri vstávaní z nízkeho sedadla. Charakterizovaná pseudohypertrofiou lýtkových svalov. Retrakcia kalkaneálnych (Achilových) šliach je menej výrazná ako pri Duchennovej chorobe.
S touto formou nie sú žiadne intelektuálne poruchy, kardiomyopatia chýba alebo je mierne vyjadrená.
Rovnako ako u iných myodystrofií viazaných na X, Beckerova forma významne zvyšuje aktivitu CPK, hoci v menšom rozsahu ako pri DMD, nepresahuje 5000 jednotiek. Gén pre Beckerovu chorobu, podobne ako Duchennova choroba, je lokalizovaný v krátkom ramene X chromozómu; je pravdepodobné, že oba lokusy spolu úzko súvisia alebo sú alelické. Na rozdiel od Duchennovej choroby, pri ktorej prakticky neexistuje dystrofia, sa pri Beckerovej chorobe syntetizuje abnormálna dystrofia. Rozdiely sa nachádzajú aj pri svalovej biopsii. Pri Beckerovej svalovej dystrofii nie sú svalové vlákna zvyčajne okrúhle, hyalínové vlákna, charakteristické pre Duchennovu svalovú dystrofiu, sú extrémne zriedkavé.
Landouzy-Dejerine myodystrofia (myodystrofia tváre a ramien). Ochorenie sa prenáša autozomálne dominantným spôsobom s vysokou penetranciou, ale trochu premenlivou expresivitou. Vyskytuje sa oveľa menej často ako Duchennova myodystrofia (0,4 na 100 tisíc obyvateľov). Predpokladá sa, že gén pre toto ochorenie je lokalizovaný na 4. chromozóme. Ženy ochorejú častejšie ako muži (3:1), Fyzické preťaženie, intenzívne športy, ako aj iracionálne fyzioterapia môže prispieť k závažnejšiemu priebehu ochorenia.
Landouzy-Dejerine myodystrofia je pomerne priaznivá súčasná forma svalovej patológie. Začína vo veku okolo 20 rokov, niekedy aj neskôr. Avšak v rodinných prípadoch ochorenia, keď je možné sledovať dynamiku mladších členov rodiny, je možné zistiť určitú slabosť svalov, napríklad svalov tváre, a v skoršom veku .
Svalová slabosť a atrofia sa najskôr prejavia v svaloch tváre resp ramenného pletenca. Postupne sa tieto poruchy šíria do svalov proximálnych ramien a potom do dolných končatín. Vo väčšine prípadov sú najskôr postihnuté svaly prednej plochy nôh (s rozvojom visiacej nohy), potom svaly proximálnych nôh. Vo vrchole ochorenia sú výrazne postihnuté kruhové svaly oka a úst, veľký prsný sval, predný pílovitý sval a dolné úseky trapézového svalu, široký chrbtový sval, biceps, triceps ramena. charakteristický vzhľad pacienti: typická tvár myopat s „priečnym úsmevom“ („úsmev La Gioconda“), výčnelok horná pera(„tapírové pery“), výrazné pterygoidné lopatky, zvláštna deformita hrudníka s jeho sploštením v predozadnom smere a rotáciou vo vnútri ramenných kĺbov. Často dochádza k asymetrii lézie, dokonca aj v rámci jedného svalu (napr. orbicularis oculi). Možno pozorovať pseudohypertrofiu gastrocnemia, deltových svalov a niekedy aj tvárových svalov. Kontraktúry a retrakcie sú vyjadrené stredne. šľachové reflexy dlho sa udržiavajú, ale niekedy v počiatočnom štádiu klesajú.
Príznaky poškodenia srdcového svalu sú zriedkavé. Aktivita sérových enzýmov je mierne zvýšená a môže byť normálna. Intelekt netrpí. Priemerná dĺžka života vo väčšine prípadov nie je znížená. Zaujímavosťou je, že EMG pri Landouzy-Dejerine myodystrofii často nie je celkom typické pre svalovú úroveň lézie. U niektorých pacientov (členov tej istej rodiny) možno pozorovať pokles amplitúdy biopotenciálov, interferenčný typ krivky, u iných naopak pokles frekvencie a hypersynchrónnej aktivity, niekedy s typickým piketom plotový rytmus. Malo by sa pamätať na spinálny variant, ktorý napodobňuje Landouzyho-Dejerineho chorobu.
Erb-Roth myodystrofia (myodystrofia končatín a pletenca). Prenáša sa autozomálne recesívnym spôsobom, obe pohlavia sú rovnako postihnuté. Začiatok ochorenia sa vo väčšine prípadov vzťahuje na polovicu 2. dekády života (14-16 rokov), popisuje sa však ako skorá, pseudo-Duchennova forma, kedy sa prvé príznaky objavia pred 10. ochorenie je ťažké a neskorý variant s nástupom po 30 rokoch.
Priebeh ochorenia môže byť rýchly alebo pomalší, v priemere k úplnej invalidite dochádza do 15-20 rokov od objavenia sa prvých príznakov. Myodystrofia začína buď poškodením svalov panvového pletenca a proximálnych nôh (forma Leiden-Mobius), alebo od ramenného pletenca (forma Erb). V niektorých prípadoch sú ramenný a panvový pletenec postihnuté súčasne. Pomerne výrazne trpia svaly chrbta a brucha. Pacienti majú charakteristickú "kačaciu" chôdzu, je ťažké vstať z ľahu a sedu, zdôraznil bedrová lordóza. Svaly tváre nie sú vo väčšine prípadov ovplyvnené. Pre túto formu nie sú charakteristické kontraktúry a pseudohypertrofia. Môžu sa vyskytnúť terminálne atrofie a retrakcie šľachy. Inteligencia je zvyčajne zachovaná. Srdcový sval je väčšinou nedotknutý. Hladina enzýmov v krvnom sére je spravidla zvýšená, ale nie tak prudko ako pri X-viazanej myodystrofii. Existujú náznaky, že u mužov je hladina CPK vyššia ako u žien. Existuje významný rozdiel v expresii mutantného génu u rôznych členov rodiny - spolu s ťažkým klinický obraz môžu byť relatívne mierne a dokonca vymazané klinické príznaky. Smrť zvyčajne nastáva v dôsledku pľúcnych komplikácií.
Keďže klinika myodystrofie končatín a pletenca je zvlášť ochotná napodobňovať nervovosvalové ochorenia iného charakteru, je potrebné najmä v ojedinelých prípadoch a pri neskorý začiatok choroby, vykonajte dôkladné klinické vyšetrenie vylúčiť spinálnu amyotrofiu, polymyozitídu, metabolické, endokrinné, toxické, medikamentózne, karcinómové myopatie. V minulosti bola jasná nadmerná diagnóza tejto formy svalovej dystrofie.
Liečba svalových dystrofií. Terapeutické možnosti svalových dystrofií sú veľmi obmedzené. Etiologické a patogenetickej liečby prakticky neexistujúce. Symptomatická liečba je zameraná predovšetkým na prevenciu vzniku kontraktúr, udržanie existujúcej svalovej sily a prípadne na určité zníženie miery atrofie. Hlavnou úlohou je maximalizovať obdobie, počas ktorého je pacient schopný samostatne sa pohybovať, pretože kontraktúry, skolióza a poruchy dýchania sa rýchlo zvyšujú v polohe na chrbte. Lekársky komplex by mala zahŕňať terapeutické cvičenia, masáže, ortopedické opatrenia, liekovú terapiu.
Terapeutická gymnastika pozostáva z pasívnych a aktívnych pohybov vykonávaných vo všetkých kĺboch v rôznych polohách: v stoji, v sede, v ľahu, s rôznymi polohami končatín. aktívne pohyby Je lepšie vykonávať v izometrickom režime. Gymnastika by sa mala vykonávať pravidelne niekoľkokrát denne. Zároveň treba varovať pred nadmerným cvičením, najmä tým, ktoré je sprevádzané presilením svalov. Dôležité (najmä po imobilizácii pacienta) sú dychové cvičenia.
Ortopedické činnosti konzervatívneho (špeciálne dlahy) a operačného charakteru (achilotómia, transekcia m. gastrocnemius), zamerané na korekciu kontraktúr a vznikajúceho patologického nastavenia končatiny, majú za cieľ aj zachovanie možnosti samostatného pohybu. V každom prípade je potrebné individuálne zvážiť očakávané prínosy a možné poškodenie z chirurgického zákroku. Treba mať na pamäti, že často (najmä pri ťažkej hyperlordóze a slabosti štvorhlavého stehenného svalu) má ekvinovárna poloha chodidiel kompenzačný význam a napríklad po achilotómii môže byť pacient úplne imobilizovaný. S rozvíjajúcimi sa kontraktúrami sa odporúča opatrné naťahovanie svalov až 20-30 krát denne, po ktorom nasleduje dlahovanie počas spánku.
Lieková terapia zahŕňa vymenovanie metabolických liekov zameraných na vyplnenie nedostatku energie a bielkovín, ale ich účinnosť je veľmi pochybná. Používajú sa antagonisty vápnika (kvôli defektu zistenému pri DMD bunkové membrányčo vedie k zvýšenému príjmu vápnika do bunky), imunomodulátory, zlúčeniny obsahujúce fosfor (ATP, fosfaden), vitamín E (100 mg perorálne 3-krát denne). Ukázalo sa, že užívanie prednizolónu (0,75 mg/kg denne) pri DMD môže dramaticky zvýšiť svalovú silu, ale tento účinok pretrváva nie dlhšie ako rok a vo všeobecnosti neovplyvňuje výsledok ochorenia. Z dôvodu vážneho vedľajšie účinky, vznikajúce vri dlhodobé užívanie drogy, jej užívanie je nevhodné. Odhady účinku anabolické steroidy kontroverzné a ich vymenovanie je často spojené s neopodstatneným rizikom. Pri hodnotení účinku niektorých liekov na DMD treba mať na pamäti, že pri strednej závažnosti ochorenia u pacientov vo veku 3-6 rokov môže dôjsť k relatívnej stabilizácii stavu súvisiacej s vekom podmieneným vývojom ochorenia. svalový systém, osvojenie si pohybových schopností, ktoré môžu do určitej miery dočasne kompenzovať prebiehajúci dystrofický proces.
Určitý význam má úprava výživy pacienta, odporúča sa diéta s vysokým obsahom bielkovín a nízkym obsahom tukov a zníženým obsahom kalórií s optimálnym obsahom vitamínov a mikroelementov. Dôležitú úlohu zohráva psychická podpora pacienta, pokračovanie vo vzdelávaní, správna profesijná orientácia.
Strana 44 zo 44
Zapojené sú kostrové svaly patologický proces s rôznymi degeneratívnymi, metabolickými a zápalové ochorenia. Vo väčšine prípadov to vedie k degenerácii svalových vlákien a pri chronických formách k ich výmene. spojivové tkanivo a tuku. Proximálne svalové skupiny sú poškodené výraznejšie ako distálne, ako aj dolné končatiny vo vzťahu k horným. Choré dieťa sa vyznačuje takzvanou kačacou (koláčavou) chôdzou, nie je schopné behať, stúpať po schodoch a vstávať, ak je v sede. Jeho šľachové reflexy sú utlmené, stupeň ich vyhasnutia je úmerný stupňu oslabenia svalovej sily. Citlivosť nie je ovplyvnená.
Medzi diagnosticky cenné laboratórne metódy patrí stanovenie aktivity enzýmov, najmä kreatínfosfokinázy, v sére. Tento enzým, ktorý katalyzuje reakciu: fosfokreatín + ADP-kreatín + ATP, je prítomný hlavne v mozgových bunkách a svalovom tkanive. Pri niektorých difúznych ochoreniach svalov, najmä svalovej dystrofii, preniká jeho nadbytočné množstvo do medzibunkového priestoru a krvi. U pacientov je aktivita sérovej laktátdehydrogenázy a glutamín oxalooctovej transaminázy zvyčajne zvýšená, ale ich široká distribúcia v iných tkanivách, vrátane pečene, znižuje špecificitu testu. Zvyčajne je na objasnenie diagnózy potrebná biopsia svalového tkaniva.
Zápalové ochorenia svalov. Zápal svalového tkaniva sprevádza niektoré infekcie, najmä trichinelózu, toxoplazmózu a tie, ktoré spôsobuje vírus Coxsackie. Často je súčasťou kolagénových ochorení, vrátane dermatomyozitídy, lupus erythematosus, periarteritis nodosa a reumatoidnej artritídy.
Polymyozitída. Difúzny izolovaný zápal svalov neznámej etiológie sa nazýva polymyozitída. Je charakterizovaný rýchlo progresívnym priebehom, slabosťou a bolesťou v proximálnych svalových skupinách. Často sú do procesu zapojené svaly krku, a preto je pre dieťa ťažké zdvihnúť hlavu a udržať ju v tejto polohe. Laboratórne príznaky zápalu svalov zahŕňajú zvýšenie ESR a počtu leukocytov. Ich absencia však nevylučuje polymyozitídu. Hladiny enzýmov v sére sú zvyčajne zvýšené. Pri svalovej biopsii sa zisťuje degenerácia a čiastočná regenerácia vlákien a ich infiltrácia lymfoidnými bunkami. Je ťažké odlíšiť polymyozitídu od svalovej dystrofie a dermatomyozitídy. Môže predstavovať atypickú formu dermatomyozitídy, hoci histológia týchto dvoch stavov je trochu odlišná: dermatomyozitída je charakterizovaná vaskulitídou, ktorá pri polymyozitíde zvyčajne chýba. Prognóza druhého z nich je o niečo priaznivejšia. Liečba kortikosteroidmi je sprevádzaná účinkom, ale pri ich zrušení môže dôjsť k relapsu.
Progresívna osifikujúca myozitída. Etiológia tohto zriedkavého ochorenia spojivového tkaniva a svalov nie je známa. Uvádza sa, že ňou trpia súrodenci vrátane dvojčiat a v priamej línii sa prenáša na pokrvných príbuzných. Predpokladá sa, že choroba sa dedí autozomálne dominantným spôsobom. Chlapci ochorejú 2-3 krát častejšie ako dievčatá.
Patologické príznaky závisia od štádia ochorenia. V počiatočných štádiách sa vo svaloch a šľachách nachádza lokálny edém a infiltráty zápalových buniek. Neskôr sú oblasti zápalu nahradené granulačným tkanivom a nakoniec sa v léziách vytvoria oblasti chrupavkového a kostného tkaniva.
Takmer 75 % chorých detí má vrodené chyby vývin, najčastejšie nevyvinutie prstov na rukách a ankylóza článkov prstov na nohách a nevyvinutie prvých prstov, polydaktýlia, zakrivenie prstov, syndaktýlia (nohy), deformácia ušnice hluchota, chýbajúce zuby. Rovnaké vrodené chyby môžu byť u príbuzných pacienta, u ktorých sa nerozvinulo progresívne ochorenie spojivového tkaniva a svalov. Vek, v ktorom sa myositis ossificans môže začať, sa líši od narodenia po staršie detstvo. Zvyčajne sa rozlišujú tri štádiá: 1) v miestach drobných lokálnych poranení vznikajú ohraničené, často teplé a na dotyk mäkké pastovité opuchy mäkkých tkanív; 2) po niekoľkých dňoch príznaky zápalu zmiznú a lézia stvrdne; 3) dochádza k osifikácii postihnutej oblasti. Pravidelne sa objavujú nové lézie, najmä na krku a chrbte. primárny príznak torticollis sa môže stať, ak sa proces vyvinul v sternocleidomastoideus sval. Nakoniec sa osifikácia rozširuje na mnohé šľachy a väzy. Nastupuje ankylóza chrbtice a kĺbov rúk a nôh (obr. 21-5). Zápal sa môže rozšíriť do temporomandibulárnych kĺbov, čo sťažuje žuvacie pohyby. Kostné výrastky môžu vyčnievať cez kožu. V dospievaní choroba často vedie k úplnej imobilizácii a smrti v dôsledku respiračné zlyhanie a zastavenie dýchania, hoci existujú správy o prípadoch prežitia. Pri osifikujúcej myozitíde existuje vysoké riziko vzniku osteogénneho sarkómu.
Ryža. 21-5. Dieťa s progresívnou myositis ossificans (typické držanie tela so stuhnutím krku a chrbta).
Niekedy je patologický proces obmedzený na miesto predchádzajúceho poranenia mäkkých tkanív (miositis ossificans circumscripta). Pri chronickej polymyozitíde a dermatomyozitíde sa môže vyskytnúť aj rozšírená kalcifikácia svalového tkaniva.
výsledky laboratórne metódyštúdie nemajú žiadnu diagnostickú hodnotu.
Hladiny vápnika, fosforu, alkalickej fosfatázy v sére, ako aj aktivita kreatínfosfokinázy a iných enzýmov zostávajú normálne. Kosť v ohnisku poškodenia sa štruktúrou nelíši od normy.
Existujúce metódy liečby sú neuspokojivé. V niektorých prípadoch sa pri užívaní ACTH a iných kortikosteroidov zaznamenalo spomalenie vývoja ochorenia. Ich úloha v konečnom výsledku liečby je otázna.
Endokrinné a metabolické myopatie. Myopatia pri hypertyreóze je pomerne zriedkavá komplikácia. Je charakterizovaná ptózou, bilaterálnou parézou tvárových svalov a svalov proximálnych končatín. Niektoré príznaky hypertyreózy môžu byť zároveň maskované svalovou slabosťou, ale tachykardiou, zvýšeným potením a zvýšeným štítna žľaza. Šľachové reflexy, na rozdiel od mnohých iných foriem myopatie, zostávajú normálne. Po úprave hypertyreózy svalová slabosť postupne mizne.
Myopatia pri hypotyreóze. Hypotyreóza u dojčiat môže byť spojená so svalovou slabosťou a hypotenziou. U starších detí s myxedémom sa svalové kontrakcie a relaxácia spomaľujú, v niektorých prípadoch je zaznamenaná svalová hypertrofia (Debre-Semelenov syndróm). Kombinácia znakov, ako je svalová slabosť a hypertrofia, naznačuje svalovú dystrofiu.
Myopatia počas liečby kortikosteroidmi. Môže skomplikovať Itsenko-Cushingovu chorobu, ale častejšie sa vyvíja pri liečbe veľkých dávok syntetických steroidov. Slabosť je badateľná najmä vo svaloch panvového pletenca, čo sa prejavuje kolísavou (kačacou) chôdzou, ťažkosťami pri výstupe do schodov a snahou vstať zo sedu. Trhnutie kolenom chýba. Môže dôjsť k rednutiu svalov. Myopatické zmeny v svalovom tkanive sú zvyčajne bezvýznamné aj pri silnej slabosti. svalovú silu po vysadení kortikosteroidov sa pomaly (do niekoľkých mesiacov) zotavuje.
Myopatia pri hyperparatyreóze. Hyperparatyreóza môže byť spojená so slabosťou a hyporeflexiou v dôsledku hyperkaliémie. Po paratyreoidektómii zvyčajne rýchlo vymiznú.
Nedostatok karnitínu (lipidová myopatia) sprevádza akumuláciu veľkého množstva lipidov vo svaloch a v dôsledku toho narušenie dodávky energie do svalov. Karnitín je jednou zo základných zložiek systému, ktorý zabezpečuje prenos mastných kyselín z dlhá reťaz z cytosolu do mitochondrií, kde podliehajú oxidácii. Svalová slabosť sa vyvíja v dvoch formách nedostatku karnitínu.
Nedostatok karnitínu vo svaloch je klinicky reprezentovaný progresívnou slabosťou ich proximálnych skupín, častejšie u školákov a dospievajúcich. Niekedy slabosť prerušovaná a kombinovaná s myoglobinúriou. V závažných prípadoch môže dôjsť k paralýze dýchacích svalov. Sérové hladiny enzýmov (kreatínkinázy a aldolázy) sú zvýšené. Elektromyogram odhaľuje nešpecifické zmeny charakteristické pre myopatiu. Vo svalovej biopsii môžete vidieť veľké množstvo kvapiek tuku. Hladina karnitínu v sére sa nemení, ale vo svaloch klesá. Rozpoznanie patológie je nevyhnutné, pretože môže byť liečiteľná. Často sa mylne považuje za svalovú dystrofiu. Účinok sa môže dostaviť po perorálnom podaní 100 mg/(kg/deň) karnitínu. V niektorých prípadoch je účinná liečba kortikosteroidmi.
- Systémový nedostatok karnitínu sa prejavuje progresívnou myopatiou, vrátane kardiomyopatie, a dysfunkciou pečene, sprevádzané klinikou hepatálnej encefalopatie, ako je Reyeov syndróm. Karnitínová insuficiencia sa od posledne menovaného líši v rekurentnom priebehu a výraznej svalovej slabosti, ktorá pretrváva medzi obdobiami exacerbácie encefalopatie. Hladina kreatínfosfokinázy v sére je výrazne zvýšená, množstvo karnitínu je znížené ako v sére, tak aj vo svaloch. Zmeny v biopsii sú podobné ako pri nedostatku karnitínu vo svalovom tkanive. Podobné klinické a morfologické zmeny, vrátane deficitu karnitínu, možno zistiť pri porušení metabolizmu organických kyselín, napríklad pri metylmalónovej a glutárovej acidúrii (sekundárny deficit karnitínu).
Ryža. 21-6. Dieťa s vrodenou absenciou ľavého prsného svalu. 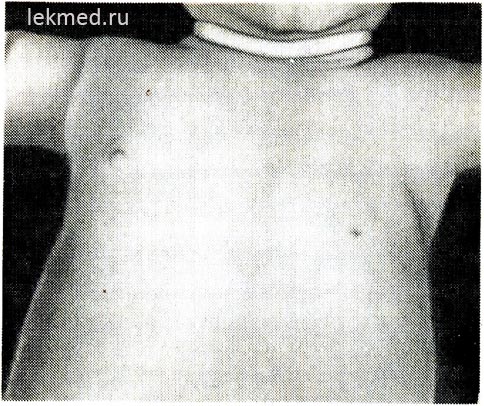
Všimnite si absenciu predného axilárneho záhybu a nízko položenej bradavky.
Liečba spočíva v udržiavaní pacienta na diéte bohatej na sacharidy a nízkym obsahom tukov a v užívaní karnitínu v dennej dávke 100 mg/kg.
Vrodené chyby svalov. Vrodená absencia svalov. Nedostatočný rozvoj svalov môže byť celkom bežný a viesť k úplnej blokáde pohybov kĺbov alebo vrodenej artrogrypóze. Ako vrodená chyba najčastejšie chýba jeden sval. Pomerne častou anomáliou je absencia sternálnej časti m. pectoralis major (obr. 21-6), v niektorých prípadoch sa tento defekt kombinuje so syndaktýliou na postihnutej strane (Poľský syndróm). Absencia prsného svalu často sprevádza svalovú dystrofiu. Vrodená absencia brušných svalov brucha je často spojená s chybami vo vývoji močových ciest.
Ryža. 21-7. Deformácia krku a asymetria tváre u chlapca s vrodenou torticollis, neliečená od 12 rokov. 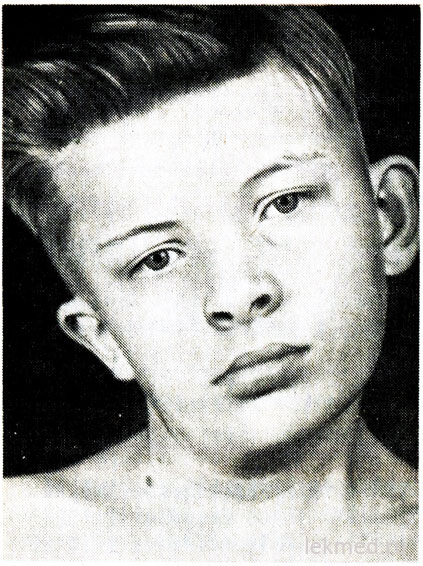
Vrodená torticollis je spôsobená jednostranným skrátením alebo kontraktúrou sternocleidomastoideus svalu. Hlava pacienta je naklonená ku kontraktúre a brada smeruje nadol v opačnom smere (obr. 21-7). Pri pokuse o korekciu polohy hlavy je cítiť výrazný svalový odpor. V postihnutom svale sú oblasti zhutnenia palpované. Príčina závady je nejasná, dlho sa považovala za následok pôrodné poranenie. Torticollis sa však vyskytuje u detí narodených chirurgicky. cisársky rez; to naznačuje, že v niektorých prípadoch sa príčina defektu týka vnútromaternicového obdobia. Torticollis treba odlíšiť od patologického záklonu hlavy v dôsledku deformácie krčných stavcov, ako je Klippel-Weilova anomália, a od zlomenín alebo dislokácií krčných stavcov. Röntgenovým vyšetrením sú vylúčené. U starších detí môže byť záklon hlavy spôsobený strabizmom, dystóniou, nádormi zadnej lebečnej jamy a cervikálny miecha, myositis ossificans, krčná lymfadenitída príp diafragmatická hernia. Vo väčšine prípadov sa dá vrodená torticollis korigovať liečebná gymnastika. Avšak, kedy chronická forma torticollis má za následok asymetrický vývoj tváre a hlavy (pozri obr. 21-7), čo si môže vyžadovať disekciu svalu na kozmetické účely.
vrodené myopatie. Do tejto skupiny patrí niekoľko zriedkavých foriem dedičných ochorení, pri ktorých sa svalová slabosť a hypotónia objavujú už od dojčenského veku (pozri tabuľku 22-1). Ich presná diagnóza je veľký význam z hľadiska predpovede. Vo všeobecnosti je priaznivá pre normálnu životnú aktivitu a dĺžku života, na rozdiel od Werdnig-Hoffmannovej choroby alebo vrodenej svalovej dystrofie. Svalová biopsia zvyčajne pomáha identifikovať vrodené myopatie.
- Ochorenie centrálneho jadra. Centrálna časť svalových vlákien je sfarbená abnormálne, ale jednotne. Vyšetrenie elektrónovým mikroskopom odhalí pokles počtu mitochondrií a vyčerpanie sarkoplazmatického retikula v centrálnej časti vlákien.
Nemalínová myopatia. Pojem "nekarmínový" sa vysvetľuje skutočnosťou, že vo svalových vláknach sú určené vláknité štruktúry.
Ryža. 21-8. Myotonická kontrakcia jazyka (a) s prudkým úderom perkusného kladiva na jeho pravú polovicu a očné viečka (b) u dieťaťa s hyperkalemickou formou familiárnej periodickej paralýzy. 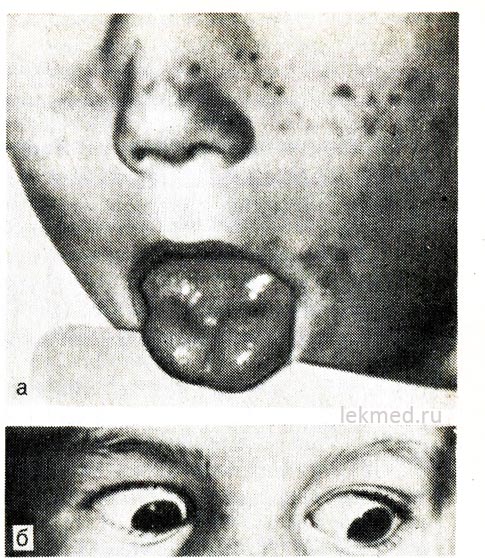
Pri pohľade nadol zostáva očné viečko stiahnuté.
Údaje z elektrónového mikroskopu naznačujú, že ide o výsledok zmien v Z-pásoch myofibríl.
Mitochondriálne myopatie. Boli hlásené niektoré formy myopatií, pri ktorých dochádza k najdôležitejším zmenám v mitochondriách svalových vlákien. Môžu sa výrazne zvýšiť ako v počte, tak vo veľkosti. Svalová slabosť a hypotónia môžu byť diagnostikované už v detstve, ale niekedy výrazne napredujú až v škole. Kardiomyopatia, encefalopatia a laktátová acidémia často sprevádzajú túto skupinu myopatií.
Myotónia. Tento stav je charakteristickým znakom rôznych svalových ochorení, ako je myotonia dystrophica, hyperkalemická familiárna paroxyzmálna paralýza a ochorenia ukladania glykogénu. Myotónia je definovaná ako výrazné oneskorenie svalovej relaxácie po dobrovoľných alebo vynútených kontrakciách. Klinicky sa prejavuje neschopnosťou uvoľniť päsť alebo viditeľnou predĺženou kontrakciou svalov po ich stimulácii, ktorá sa prejavuje prudkým podráždením (obr. 21-8). Dá sa to pozorovať, ak perkusným kladivom zasiahnete povrchovú skupinu svalov, napríklad svaly jazyka alebo dlaňovú plochu v oblasti elevácie prvého prsta. Myotónia je potvrdená elektromyografickými údajmi. V tomto prípade je charakteristická spontánna aktivita svalov badateľná po ich uvoľnení alebo dobrovoľnej kontrakcii (myotonické výboje).
Myotonia congenita (Thomsenova choroba). Jediným znakom tohto ochorenia, zdedeného dominantným typom, je myotónia. Môže sa prejaviť v dojčenskom veku v podobe spomalenia prehĺtacích pohybov a následného zvracania
účinok neschopnosti normálnej relaxácie svalov hltanu. V staršom detstva myotónia sa prejavuje ako neschopnosť pacienta uvoľniť prsty zovreté v päsť. Pri prvom pokuse o vykonanie nejakého pohybu svaly dieťaťa stvrdnú. Opakovaným opakovaním toho istého pohybu sa trochu uvoľnia. Takže napríklad choré dieťa zažíva veľké ťažkosti na začiatku aktu chôdze. Prvé kroky zvyčajne robí veľmi váhavo a pomaly. Po niekoľkých sekundách sa chôdza stáva normálnou alebo takmer normálnou. Symptómy myotónie sa zhoršujú nepriaznivým emočným stavom pacienta a ochladzovaním tela. Svalová sila zostáva v norme, svaly sú dostatočne vyvinuté a často nápadne zväčšené, čo vytvára mylný dojem o atletickej konštitúcii pacienta.
Diagnóza je založená na klinických nálezoch a elektromyografických údajoch. Aktivita sérových enzýmov je v rámci normálnych limitov. Jediným histologickým znakom je hypertrofia svalových vlákien.
Ochorenie sa líši od dystrofickej myotónie absenciou svalovej slabosti a atrofie a dystrofických zmien v biopsii svalového tkaniva. Liečba novokaínom alebo chinidínsulfátom je sprevádzaná účinkom a je indikovaná na funkčné poruchy. Priebeh ochorenia je zvyčajne benígny, s vekom sa môže stav pacienta zlepšovať.
paroxyzmálna paralýza. Táto skupina ochorení je charakterizovaná periodickou svalovou slabosťou s úplnou alebo takmer úplnou obnovou svalovej sily v období medzi záchvatmi. Zahŕňa tiež nedostatok svalovej fosforylázy (McArdleova choroba).
Hyperkalemická paroxyzmálna paralýza. Dedičná epizodická adynamia alebo paramyotónia sa prenáša podľa dominantného typu a je obzvlášť závažná u mužov. Zvyčajne začína v ranom detstve (niekedy v detstve). Útoky sa vyskytujú počas obdobia odpočinku po veľkom zaťažení svalov. Slabosť sa vyvíja rýchlo a môže trvať niekoľko hodín. Zvlášť sa to cíti v nohách; respiračná funkcia zvyčajne nie je narušená. Často je adynamia sprevádzaná myotóniou, ktorá pretrváva medzi záchvatmi, čo sa najzreteľnejšie prejavuje vo forme oneskorenia pohybu očných viečok pri pohľade nadol (pozri obr. 21-8, b).
Hladiny draslíka v sére sú často zvýšené počas záchvatu, ale na presné určenie môžu byť potrebné viaceré štúdie počas niekoľkých záchvatov. Útok je možné umelo vyvolať pomocou draslíkovej záťaže (2-3 g perorálne), ale mal by sa vykonávať iba pod kontrolou EKG. Opakované útoky zastaví diakarb. Ťažké formy ochorenia sú charakterizované rozvojom chronickej, miernej slabosti a degeneratívnych zmien vo svaloch.
Hypokaliemická paroxyzmálna paralýza. Rodinná záchvatová paralýza, tiež zdedená podľa dominantného typu, je obzvlášť náročná u chlapcov. Na rozdiel od hyperkalemickej formy sa prvý záchvat objavuje v neskorom detstve alebo v ranej adolescencii. Dôvodom je konzumácia bohatého jedla bohatého na sacharidy alebo odpočinok po ňom fyzická aktivita. Útok zvyčajne začína na ďalšie ráno po ťažkej fyzickej námahe a ťažkom jedle. Je charakterizovaná svalovou slabosťou a areflexiou. Môže byť narušená funkcia dýchania. Môže sa pripojiť arytmia, vrátane ventrikulárneho extrasystolu a tachykardie. Záchvaty môžu trvať aj viac ako 24 hodín.V paralytickej fáze hladina draslíka v sére zvyčajne klesá (2-3 mmol/l). Základná chyba nie je známa. U pacientov s opakujúcimi sa ťažkými záchvatmi sa rozvinie chronická svalová slabosť a patologické zmeny vo svaloch. Liečba počas záchvatov spočíva v užívaní chloridu draselného; jeho počiatočná dávka je 2-3 g Diakarb pomáha znižovať frekvenciu záchvatov.
Paroxyzmálna myoglobinúria (idiopatická myoglobinúria). Idiopatická myoglobinúria je heterogénna skupina porúch, pri ktorých sa záchvaty paralýzy s myoglobinúriou vyskytujú spontánne alebo po intenzívnom cvičení. Choroba sa dedí dominantným spôsobom, spojený s chromozómom X. Svaly, najčastejšie lýtka a stehná, sú pri záchvate bolestivé a opuchnuté. Moč sa stáva tmavočerveným resp Hnedá farba. Myoglobinúria môže spôsobiť renálnu tubulárnu nekrózu, ktorá je smrteľná zlyhanie obličiek.
Diagnóza je potvrdená detekciou myoglobulínu v moči. Pozitívny benzidínový test v neprítomnosti erytrocytov v moči potvrdzuje prítomnosť myoglobínu v ňom, najmä ak hemoglobín nie je zistený v sére. Hemoglobín sa stanovuje spektrofotometriou. Paroxyzmálnu myoglobinúriu treba odlíšiť od McArdleovej choroby, deficitu karnitín palmityltransferázy a myoglobinúrie po nezvyčajnom namáhavom cvičení alebo svalovom poranení zdravý človek. Myoglobinúria po ťažkom svalovom cvičení sa vyskytuje pri pseudohypertrofickej svalovej dystrofii (Duchennova choroba).
Liečba pozostáva z odpočinku na lôžku; v prípade potreby vykonať umelé vetranie pľúca. Aby sa zabránilo zlyhaniu obličiek, je potrebné pacientovi predpísať bohatý nápoj.
Nedostatok karnitín palmityltransferázy. Pri nedostatku tohto enzýmu je narušený prenos mastných kyselín s dlhým reťazcom do mitochondriálnych segmentov, v ktorých prebieha oxidácia a tvorba ketónov. Nedostatok izoenzýmu typu II sa dedí recesívnym spôsobom. V dôsledku jeho nedostatku je narušená ketogenéza v tkanivách vrátane svalov a pečene. Prvé príznaky ochorenia sa objavujú častejšie u detí v škole a dospievaní. Pozostávajú z opakovaných epizód svalovej bolesti, slabosti a horúčky po cvičení alebo nalačno. Myoglobinúria sprevádzajúca záchvaty môže viesť k zlyhaniu obličiek. Pôst vedie k hypoglykémii. Medzi útokmi sa deti javia ako zdravé. Ochorenie sa musí odlíšiť od iných stavov sprevádzaných periodickou slabosťou a myoglobinúriou. Metóda stanovenia aktivity karnitín palmityltransferázy má diferenciálnu diagnostickú hodnotu. Klesá vo svaloch a pečeňové tkanivá leukocyty a fibroblastová kultúra. Konzumácia stravy bohatej na sacharidy s nízkym obsahom tuku môže pomôcť znížiť záchvaty.
Svalové dystrofie. Tieto anomálie patria do skupiny rodinných ochorení sprevádzaných degeneráciou svalových vlákien. Klasifikácia svalových dystrofií je založená na znakoch, ako je čas nástupu, rýchlosť progresie, distribúcia lézií podľa svalovej skupiny a spôsob dedičnosti.
Pseudohypertrofická svalová dystrofia. Detstvo alebo Duchennova forma je najbežnejšou formou svalovej dystrofie; jeho frekvencia je 0,14 na 1000 detí. V klasickej forme sa vyskytuje len u chlapcov a dedičnosť viazaná na X chromozóm sa vyskytuje približne u 50 % probandov. V iných prípadoch je choroba spôsobená novými mutáciami. Uvádza sa zriedkavá forma svalovej dystrofie, klinicky identická s Duchennovou formou, ale dedičná recesívnym typom s rovnakou frekvenciou ochorenia u chlapcov a dievčat. Spoľahlivo diagnostikovať ochorenie je zriedka možné u dieťaťa mladšieho ako 3 roky. Anamnéza zvyčajne naznačuje, že dieťa malo oneskorený vývoj motorické funkcie, začal neskoro sedieť, chodiť a behať, čo, samozrejme, naznačuje viac skorý štart choroby. Častá je kolísavá (kačacia) chôdza, ťažkosti pri chôdzi po schodoch, hypertrofia lýtkových svalov. klinické prejavy. V niektorých prípadoch sú do procesu zapojené aj iné svaly, najmä deltový sval, brachioradialis a svaly jazyka.
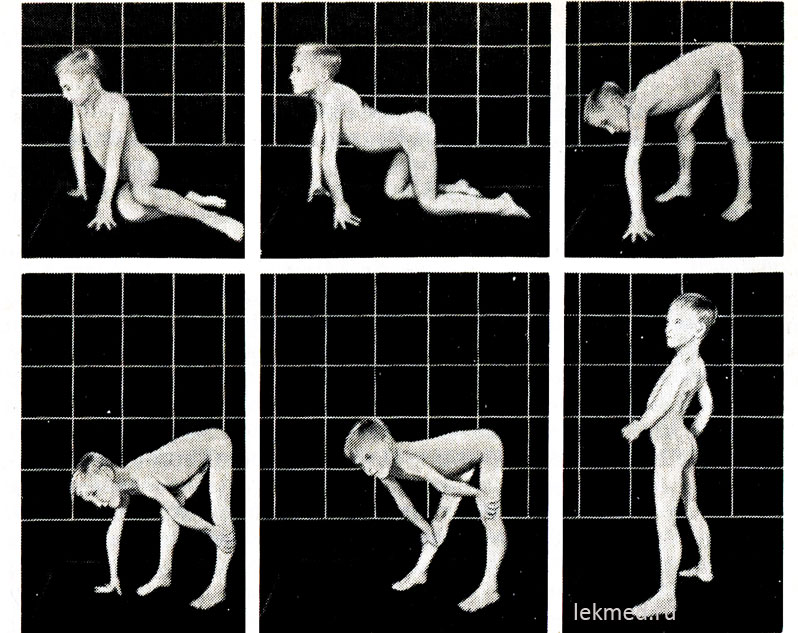
Ryža. 21-9. Typické polohy zaujaté pri vstávaní z podlahy (znak Govers) u 7-ročného dieťaťa s pseudohypertrofickou myopatiou.
V stoji (posledná fotografia) je výrazne výrazná lordóza.
Na začiatku ochorenia majú hypertrofované svaly výraznú silu, ale neskôr sa znižuje (pseudohypertrofia), pretože k nárastu svalovej hmoty dochádza v dôsledku ich tukovej infiltrácie. Sila hypertrofovaného svalu gastrocnemia výrazne prevyšuje silu svalov prednej plochy nohy, čo vysvetľuje časté kontraktúry pätovej šľachy a chod dieťaťa po prstoch. Slabosť svalov panvového pletenca sa prejavuje charakteristickou kačacou (pánovou) chôdzou a ťažkosťami, ktoré dieťa zažíva, keď vstáva zo sedu na podlahe. Pri dostatočne ťažkých formách svalovej dystrofie má dieťa Goversov príznak: vstáva z podlahy, najprv si kľakne, opiera sa o ruky a potom vstáva, pričom postupne tlačí ruky z holene, kolenných kĺbov a stehná (obr. 21-9). Slabosť svalov ramenného pletenca zistíte tak, že dieťa držíte vo zvýšenej polohe za podpazušie. Normálne sa snaží udržať pritlačením rúk k telu; pri svalovej dystrofii akoby prekĺzla cez ruky skúšajúceho. Choré dieťa často nedokáže zdvihnúť ruky nad hlavu. V neskorších štádiách ochorenia vzniká výrazná svalová atrofia. Zvyčajne vo veku 12 rokov už dieťa nemôže chodiť. Pacienti v 75% prípadov zomierajú pred dosiahnutím veku 20 rokov. Väčšina z nich má kardiomyopatiu, ktorá v niektorých prípadoch spôsobuje neočakávaná smrť. Ak je dedičnosť viazaná na X a choroba sa začala v staršom detstve, očakávaná dĺžka života zostáva dlhá (Beckerova svalová dystrofia). Priemerné IQ detí s DMD je 80; 25% detí má mentálnu retardáciu.
O odlišná diagnóza Duchennova svalová dystrofia by mala zahŕňať Werdnigovu-Hoffmannovu chorobu u starších dojčiat a svalové choroby, ako sú endokrinné myopatie, nedostatok karnitínu, choroby ukladania glykogénu a polymyozitída. Niekedy pri kontraktúrach šľachy kalkanea a chôdzi dieťaťa po prstoch na nohách možno predpokladať detskú mozgovú obrnu, ale pri svalovej dystrofii nie sú charakteristické znaky mozgová obrna spasticita a hyperreflexia.
Diagnóza je založená na stanovení aktivity enzýmov v sére, elektromyografických údajoch a biopsii svalového tkaniva. Aktivita enzýmov, najmä kreatínfosfokinázy, ešte pred vývojom klinické príznakyčasto prekračuje normu 10-krát aj u dojčiat. Na elektromyograme sa najprv odhalí zníženie trvania a zníženie amplitúdy motorických potenciálov. Histologické zmeny pozostávajú z degenerácie svalových vlákien. Často sa líšia veľkosťou a sú čiastočne nahradené tukom a spojivovým tkanivom. Veľkosť ich jadier sa tiež líši. Diagnózu možno stanoviť pri narodení stanovením aktivity kreatínfosfokinázy. Metódy identifikácie prenášačiek ešte neboli vyvinuté, napriek tomu, že 60 – 80 % z nich vykazuje mierny alebo mierny nárast jeho hladiny. Tieto znaky sú typické skôr pre detstvo ako pre nasledujúce obdobia života.
účinných metód neexistuje žiadny liek. Pacient by mal byť aktívny a mal by byť schopný čo najviac chodiť. Je potrebné zabezpečiť, aby sa dieťa vyhýbalo intenzívnej fyzickej aktivite, pretože môže spôsobiť pretrhnutie svalových vlákien. V niektorých prípadoch chirurgické predĺženie kalkaneálnej šľachy zlepšuje schopnosť chôdze, ale predlžuje sa pokoj na lôžku po ortopedickej korekcii sa môže zvýšiť svalová atrofia. Genetické poradenstvo zohráva dôležitú úlohu.
Vrodená svalová dystrofia. Ochorenie sa dedí autozomálne recesívnym spôsobom a je charakterizované svalovou hypotenziou a slabosťou u dojčiat. Je zaradený do skupiny stavov definovaných ako „lenivé dieťa“ (pozri tabuľku 21-1). Nástup ochorenia sa vzťahuje na vnútromaternicové obdobie. Niekedy má novorodenec výraznú atrofiu svalov, ich kontraktúry, obmedzenú pohyblivosť kĺbov. Odlíšenie od Werdnig-Hoffmannovej choroby je ťažké. Fascikulácie jazyka, charakteristické pre druhú, chýbajú pri svalovej dystrofii. Šľachové reflexy sú potlačené, ale nie úplne stratené. Tento proces zahŕňa svaly zapojené do dýchania vrátane bránice. V závažných prípadoch nastáva smrť pred dosiahnutím veku 1 roka v dôsledku zlyhania dýchania; pri ľahších formách sa dlhodobo udržiava normálna životaschopnosť. Zvýšenie aktivity sérových enzýmov nie je zaznamenané, hoci vo svaloch sa vyskytujú dystrofické zmeny.
Rameno-tvárová forma svalovej dystrofie. To stačí mierna forma svalová dystrofia sa dedí autozomálne dominantným spôsobom. Zvyčajne začína vo veku 10-20 rokov a je charakterizovaná slabosťou a atrofiou svalov tváre a ramenného pletenca. Tvár je úplne amimická, pacient nemôže zavrieť oči a zapískať. Choroba postupuje pomaly a je kompatibilná s normálnou dĺžkou života. Diagnóza je založená na klinickom náleze a type dedičnosti. Výsledky biopsie svalového tkaniva naznačujú dystrofické zmeny v ňom. Hladiny kreatínfosfokinázy v sére môžu zostať normálne alebo mierne zvýšené.
Panvová forma svalovej dystrofie. Táto skupina heterogénnych porúch je charakterizovaná pomalou progresiou svalovej dystrofie a je dedená autozomálne recesívnym spôsobom. Začiatok ochorenia sa vzťahuje na staršie detstvo, dospievanie alebo dospelosť. Zvyčajne sú postihnuté svaly panvového pletenca.
Očná forma myopatie. Dystrofické zmeny sa vyskytujú najmä vo vonkajších očných svaloch. Choroba začína v detstve alebo dospievaní. S ním napreduje ptóza a obmedzenie pohybov. očné buľvy. Niekedy sa slabosť rozširuje na svaly tváre a krku. Ochorenie treba odlíšiť od myasthenia gravis a paralýzy hlavových nervov s nádormi mozgového kmeňa.
Progresívna oftalmoplégia, začínajúca v detstve alebo dospievaní, je spojená s atypickou retinitis pigmentosa a srdcovým blokom (Kearns-Sayersov syndróm). Zvyčajne sa spája s progresívnou ataxiou, oneskoreným rastom a pubertou. Pod sarkolemou svalov sa určujú veľké akumulácie atypických mitochondrií. Genetická povaha tohto procesu nebola stanovená. Pomocou kardiostimulátora môžete kontrolovať možnosť náhlej smrti v dôsledku porušenia srdcového vedenia.
Myotonická dystrofia. Napriek tomu, že myotonická dystrofia začína akoby u dospelého človeka, jej nástup sa čoraz častejšie zaznamenáva u dojčiat a neskôr u detí. Dedí sa autozomálne dominantným spôsobom. Jeho nástup v detstve naznačuje, že matka trpí myotóniou. V súlade s tým môžu vnútromaternicové faktory ovplyvniť závažnosť ochorenia u dieťaťa. Už v čase narodenia vie určiť svalová hypotenzia chýba mu schopnosť sať. Zaostávanie fyzického a duševného vývoja sa zvyčajne zistí neskôr. V ranom detstve sa svalová slabosť a atrofia rozšírili hlavne na tvárové, čeľustné a temporálne svaly. Zvyčajne sa zaznamenáva bilaterálna ptóza. Medzi diagnosticky významné metódy patrí svalová perkusia, elektromyografia; pre týchto pacientov je typická neschopnosť uvoľniť ruku zovretú v päsť (pozri Vrodená myotónia). Slabosť a atrofia svalov končatín a panvového pletenca (zvyčajne distálne skupiny) sa zisťujú vo vyššom detstve alebo dospievaní. U dospelých je toto ochorenie sprevádzané šedým zákalom, plešatosťou, atrofiou semenníkov.
Diagnóza je založená na identifikácii príznakov myotónie, charakteristickej distribúcie svalovej slabosti, dedičnosti podľa dominantného typu a dystrofických zmien vo svaloch. V detskom veku môže byť priebeh ochorenia nepriaznivý, často sprevádzaný mentálna retardácia. V období dospievania sa do popredia dostáva svalová slabosť. Pri funkčných poruchách je indikovaná liečba novokaínom a chinidínom.

