Amyotrofická laterálna skleróza (ALS), známa aj ako Lou Gehrigova choroba a Charcotova choroba, špecifické ochoreniečo vedie k bunkovej smrti. V Spojenom kráľovstve sa používa pojem choroba motorických neurónov (MND), zatiaľ čo v iných krajinách sa tento pojem používa na 5 typov chorôb, z ktorých je najbežnejšia ALS. Je charakterizovaná svalovou atrofiou, svalovými kŕčmi a postupne sa zvyšujúcou slabosťou v dôsledku straty svalové tkanivo. To vedie k ťažkostiam s rozprávaním, prehĺtaním a v niektorých prípadoch aj s dýchaním. Príčina je neznáma v 90-95% prípadov. Asi 5-10% prípadov je dedičných. Približne polovica prípadov je spôsobená jedným alebo dvoma špecifickými génmi. To vedie k smrti neurónov, ktoré riadia dobrovoľné svalové kontrakcie. Diagnóza je založená na príznakoch a symptómoch po testovaní, aby sa vylúčili ďalšie potenciálne stavy. Neexistuje žiadny liek na ALS. Liek nazývaný riluzol spomaľuje progresiu ALS a zvyšuje očakávanú dĺžku života o 2-3 mesiace. Neinvazívna ventilácia môže zlepšiť stav pacienta a dĺžku života. Ochorenie sa zvyčajne rozvíja od 60. roku života, v príp dedičné ochorenie– od 50. Priemerná dĺžka života od začiatku ochorenia je 3-4 roky. Približne 10 % pacientov žije viac ako 10 rokov. Väčšina zomiera na zástavu dýchania. Vo väčšine krajín nie je výskyt ALS známy. V Európe a USA sa toto ochorenie vyskytuje u 2 zo 100 000 ľudí ročne. Chorobu prvýkrát opísal Charles Bell v roku 1824. Vzťah medzi symptómami a základnými neurologickými problémami opísal Jean-Martin Charcot v roku 1869 a v roku 1874 začal používať samotný termín „amyotrofická laterálna skleróza“. V USA sa choroba stala známou po tom, čo zasiahla slávneho bejzbalového hráča Lou Gehriga a Stephen Hawking sa preslávil svojimi vedeckými úspechmi. V roku 2014 bola spustená online kampaň na zvýšenie povedomia o ALS. V rámci tejto kampane s názvom ice bucket challenge (doslova - test s vedrom ľadovej vody) sa ľudia oblievali vedrom vody a prispievali.
príznaky a symptómy
Ochorenie spôsobuje slabosť a chradnutie svalov v celom tele v dôsledku degenerácie horných a dolných motorických neurónov. Postihnutí jedinci môžu nakoniec stratiť schopnosť ovládať vôľové pohyby, ale močový mechúr, črevá a svaly pohybu očí fungujú až do pokročilých štádií ochorenia. Kognitívne funkcie sú u väčšiny ľudí tiež zachované, hoci u niektorých (asi 5 %) sa vyvinie frontotemporálna demencia. U mnohých (30–50 %) sa vyskytujú aj menšie kognitívne zmeny, ktoré môžu zostať nepovšimnuté, ale sú viditeľné pri podrobnom neuropsychologickom testovaní. ALS sa zriedkavo vyskytuje u ľudí s degeneratívnym ochorením svalov a kostí (súčasť syndrómu multisystémovej proteinopatie). Ochorenie nepostihuje zmyslové nervy a autonómny nervový systém, čo znamená, že väčšina ľudí si zachováva sluch, zrak, citlivosť, čuch a chuť.
Primárne príznaky
Skoré príznaky ochorenia sú celkom všeobecné (slabosť a/alebo svalová atrofia), takže diagnostika je ťažká. Ďalšie príznaky zahŕňajú ťažkosti s prehĺtaním, kŕče alebo stuhnutosť postihnutých svalov, svalovú slabosť (postihujúcu ruky alebo nohy) a/alebo nezrozumiteľnú reč a nosové priechody. V prvých štádiách ochorenia je postihnutá tá časť tela v závislosti od toho, ktoré motorické neuróny sú poškodené ako prvé. Asi 75 % ľudí s týmto ochorením najprv pociťuje slabosť alebo atrofiu rúk alebo nôh. Pri chôdzi alebo behu dochádza k nemotornosti, človek môže zakopnúť alebo zakopnúť, prípadne ťahať nohu kúsok za sebou (syndróm padnutej nohy). Ak je postihnutá ruka, dochádza k ťažkostiam pri pohyboch, ktoré si vyžadujú manuálnu zručnosť (napríklad zapínanie gombíkov na košeli, písanie niečoho, otáčanie kľúčom v zámku). Symptómy môžu pretrvávať iba v jednej končatine po dlhú dobu alebo počas celej choroby; príznak je známy ako amyotrofia jednej končatiny. Asi 25% všetkých chorôb začína ako progresívny bulbárny syndróm. Primárne príznaky sa v tomto prípade prejavujú ťažkosťami s rečou alebo prehĺtaním. Reč sa stáva zmätenou, tichšou a nosovejšou. Ťažkosti s prehĺtaním sú sprevádzané stratou pohyblivosti jazyka. U malého počtu ľudí sú ako prvé postihnuté medzirebrové svaly, ktoré prispievajú k dýchaciemu procesu. Malé percento ľudí má frontotemporálnu demenciu, ale s progresiou ochorenia sa objavujú typické príznaky ALS. V priebehu času majú ľudia ťažkosti s pohybom, prehĺtaním (dysfágia), rozprávaním a tvorením slov (dyzartria). Dysfunkcia horných motorických neurónov sa prejavuje svalovou tvrdosťou (spasticitou) a zvýšenou reflexná aktivita(hyperreflexia), ako aj hyperaktívny dávivý reflex. Babinského reflex tiež naznačuje poškodenie horného motorického neurónu. Symptómy poškodenia dolného motorického neurónu sú svalová slabosť a atrofia, svalové kŕče, ktoré možno vidieť pod kožou, ale malé kŕče nie sú diagnostikovateľným príznakom, pozorujú sa neskôr alebo sprevádzajú slabosť a atrofiu. Asi 15 – 45 % ľudí pociťuje afektívnu labilitu, neurologickú poruchu známu ako emočná nestabilita, ktorá pozostáva zo záchvatov nekontrolovateľného smiechu, plaču, neustáleho úsmevu, čo je spojené s degeneráciou bulbárnych horných motorických neurónov, ktorá sa prejavuje nadmerným prejavom emócií. . Aby bola diagnostikovaná ALS, osoba musí mať príznaky poškodenia horných a dolných neurónov, ktoré nie sú vlastné iným ochoreniam.
Vývoj choroby
Aj keď sa poradie a rýchlosť symptómov u jednotlivcov líši, väčšina ľudí nie je schopná chodiť ani používať ruky. Strácajú tiež schopnosť rozprávať a prehĺtať jedlo, preto sa najčastejšie používa neinvazívna ventilácia BiPAP (BiPAP). Rýchlosť progresie ochorenia sa hodnotí pomocou škály ALSFRS-R, čo je dotazník alebo klinický rozhovor so stupnicou od 48 (normálne fungovanie) do 0 (ťažká forma). Hoci je miera variácie vysoká a malý počet ľudí má nízky stupeň ochorenia, v priemere pacienti strácajú 0,9 bodu za mesiac. Štúdia založená na klinických vyšetreniach ukázala 20 % zmenu v ALSFRS-R ako klinicky významnú. Bez ohľadu na to, ktorá časť tela je postihnutá ako prvá, choroba sa vo svojom vývoji šíri do iných častí tela. Symptómy končatín sa zvyčajne rozšíria skôr na protiľahlé končatiny ako do novej časti tela, zatiaľ čo bulbárny začiatok ochorenia najskôr postihuje ruky a potom nohy. Rýchlosť progresie ochorenia je nižšia u pacientov mladších ako 40 rokov na začiatku, u pacientov s nízkou úrovňou obezity, u jedincov s ochorením jednej končatiny a u pacientov s primárnymi príznakmi ochorenia horného motorického neurónu. Naopak, rýchlosť progresie ochorenia je vyššia a prognóza horšia u ľudí s bulbárnou, respiračnou a frontotemporálnou demenciou. Alelický variant CX3CR1 má vplyv na progresiu ochorenia a očakávanú dĺžku života.
Neskoré štádiá
Hoci asistovaná ventilácia môže zmierniť problémy s dýchaním a predĺžiť očakávanú dĺžku života, nespomalí progresiu ALS. Väčšina ľudí s ALS zomiera na zástavu dýchania do 3-5 rokov od nástupu symptómov. Priemerná dĺžka života od prepuknutia choroby po smrť je 39 mesiacov a len 4 % ľudí žije dlhšie ako 10 rokov. Gitarista Jason Becker žije s touto chorobou od roku 1989 a fyzik Stephen Hawking žije s touto chorobou viac ako 50 rokov, ale všetky tieto prípady sú jedinečné. Ťažkosti pri žuvaní a prehĺtaní sťažujú jedenie a zvyšujú riziko zadusenia a požitia potravy do pľúc. V neskorších štádiách ochorenia sa môže vyvinúť aspiračná pneumónia a udržiavanie hmotnosti sa môže stať vážnym problémom, ktorý si môže vyžadovať sondu na kŕmenie. S oslabením bránice a medzirebrových svalov hrudník, ktoré podporujú dýchanie, funkciu pľúc, či skôr vitálna kapacita a inspiračný tlak, klesá. Pri respiračnej forme ochorenia k tomu môže dôjsť aj pred oslabením svalstva končatín. Väčšina pacientov s ALS zomiera na zástavu dýchania alebo zápal pľúc. Zapnuté záverečné fázy ochorenie môže byť ovplyvnené okulomotorický nerv, ktorý riadi pohyb, podobne ako svaly očnej gule. Pohyb očí je udržiavaný až do posledných štádií kvôli svalovým rozdielom očná buľva s kostrovým svalstvom, ktoré sú ako prvé postihnuté chorobou. V neskorších štádiách ochorenia môže stav pacienta pripomínať locked-in syndróm.
pohyb očí
Ľudia s ALS môžu mať problémy s rýchlymi dobrovoľnými pohybmi očí. Rýchlosť pohybu očí sa spomaľuje. Existuje aj kŕč konvergencie očí. Testovanie vestibulo-očného reflexu môže pomôcť identifikovať tieto problémy. Elektrookulografia (EOG) meria pokojový potenciál sietnice. U ľudí s ALS EOG demonštruje zmeny, ktoré ovplyvňujú progresiu ochorenia a tiež poskytuje údaje pre klinické hodnotenie progresie ochorenia, ktoré ovplyvňuje okulomotorickú aktivitu. Okrem toho vám EOG umožňuje identifikovať problémy s očami v počiatočnom štádiu. Okulomotorické svaly sa líšia od ostatných kostrových svalov. Okulomotorický sval je jedinečný v tom, že sa počas života neustále mení a počas starnutia si udržiava počet satelitných buniek. V okohybných svaloch je oveľa viac myogénnych progenitorových buniek ako v kostrových svaloch končatín.
Príčiny
genetika
V 5-10% prípadov je choroba priamo zdedená od rodičov. Asi 20 % rodinných prípadov (alebo 2 % všetkých prípadov) je spojených s mutáciou na chromozóme 21, ktorý kóduje superoxiddismutázu. Existuje viac ako sto foriem mutácií. IN Severná Amerika najbežnejším mutantným génom je mutantný gén SOD1; vyznačuje sa neuveriteľne vysokou mierou progresie od času choroby do času smrti. Najbežnejším mutantom v škandinávskych krajinách je D90A-SOD1 s pomalšou rýchlosťou progresie ako typická ALS a ľudia s touto formou ochorenia žijú v priemere 11 rokov. V roku 2011 bola v C9orf72 objavená genetická anomália známa ako hexanukleotidová repetícia, táto anomália je spojená s ALS a frontotemporálnou demenciou a predstavuje až 6 % všetkých prípadov ALS medzi bielymi Európanmi. Tento gén majú aj ľudia filipínskeho pôvodu. Gén UBQLN2 je zodpovedný za produkciu proteínu ubiquilín 2 v bunke, ktorý je členom rodiny ubiquilínov a riadi degradáciu ubiquitinovaných proteínov. Mutácie v UBQLN2 zabraňujú degradácii proteínov, čo vedie k neurodegenerácii a spôsobuje (prevažne dedičnú) X-viazanú ALS a ALS/demenciu.
SOD1
V roku 1993 vedci zistili, že mutácia v géne (SOD1), ktorý produkuje enzým Cu-Zn superoxiddismutáza (SOD1), je spojená s 20 % prípadov dedičnej ALS. Tento enzým je pomerne silný antioxidant, ktorý chráni telo pred poškodením spôsobeným superoxidom, toxickým voľným radikálom generovaným v mitochondriách. Voľné radikály sú vysoko reaktívne molekuly produkované bunkami počas metabolizmu. Voľné radikály sa môžu hromadiť a spôsobiť poškodenie DNA a proteínov v bunke. V súčasnosti je s ALS spojených viac ako 110 rôznych mutácií v SOD1, z ktorých niektoré (napríklad H46R) majú veľmi dlhú klinickú históriu, zatiaľ čo iné, ako napríklad A4V, sú mimoriadne agresívne. Pri oslabení obrany proti oxidačnému stresu sa aktivuje bunková smrť (apoptóza). Porucha SOD1 môže viesť k strate alebo získaniu funkčnosti. Strata funkcie SOD1 môže viesť k akumulácii voľných radikálov. Získanie funkcií SOD1 môže byť toxické. V dôsledku štúdií zahŕňajúcich transgénne myši bolo navrhnutých niekoľko teórií o úlohe SOD1 v dedičnej ALS. U myší, ktorým chýba gén SOD1, sa ALS nevyvinie, aj keď sa u nich prejaví zrýchlená svalová atrofia súvisiaca s vekom (sarkopénia) a znížená dĺžka života. To znamená, že toxické vlastnosti mutácie SOD1 sú výsledkom zisku funkcie, nie straty. Okrem toho sa zistilo, že akumulácia proteínov je patologickým znakom dedičnej a sporadickej ALS (proteinopatie). Je zvláštne, že u myší s mutovanou SOD1 (najčastejšie mutáciou G93A) sa akumulácia (chybne poskladaný proteín) mutovaného SOD1 zistila iba v postihnutých tkanivách, pričom väčšia akumulácia sa pozorovala počas degenerácie motorických neurónov. Vedci sa domnievajú, že akumulácia mutovaného SOD1 hrá dôležitú úlohu pri narušení bunkových funkcií prostredníctvom poškodenia mitochondrií, proteazómu, chaperónov a iných proteínov. Ak sa potvrdí, ktorákoľvek z týchto abnormalít môže slúžiť ako dôkaz, že takéto akumulácie vedú k toxicite mutovaného mutovaného SOD1. Kritici poukazujú na to, že u ľudí spôsobujú mutácie SOD1 iba 2 % všetkých prípadov ochorenia a kauzálne mechanizmy sa môžu líšiť od mechanizmov zodpovedných za sporadickú formu ochorenia. V súčasnosti zostávajú myši ALS-SOD1 najlepším modelom ochorenia v predklinických štúdiách, ale je nádej, že sa vyvinie nový model. K dispozícii je online databáza, čo je vedecká komunita a platforma s aktuálnymi informáciami o ALS pre širokú verejnosť. Stránka sa volá ALSOD, pôvodne bola vytvorená pre publikácie o géne SOD1 v roku 1999, v súčasnosti sa na stránke nachádza viac ako 40 génov súvisiacich s ALS.
Iné faktory
V prípade, že ochorenie nie je dedičné, teda v 90 % prípadov sú príčiny ochorenia neznáme. Možné dôvody aj keď sú nespoľahlivé, sú to úrazy hlavy, vojenská služba, časté užívanie drog a účasť na kontaktných športoch. Výskum sa zameriava aj na úlohu glutamátu pri degenerácii motorických neurónov. Glutamát je jedným z neurotransmiterov v mozgu. Vedci zistili, že ľudia s ALS majú v porovnaní so zdravými viac vysoký stupeň glutamátu v krvi a mozgovomiechovom moku. Riluzol je v súčasnosti jediným liekom schváleným v USA na liečbu ALS a účinkov na transportéry glutamátu. Droga má nízky účinok na predlžovanie dĺžky života, čo naznačuje, že nadbytok glutamátu nie je jedinou príčinou ochorenia. Niektoré výskumy naznačujú súvislosť medzi sporadickým ALS (najmä u športovcov) a stravou bohatou na aminokyseliny s rozvetveným reťazcom (populárny doplnok medzi športovcami), ktoré spôsobujú bunkovú excitabilitu podobnú tej u ľudí s ALS. Bunková excitabilita vedie k zvýšenej absorpcii vápnika bunkou, čo vedie k smrti neurónových buniek. Niektoré dôkazy naznačujú, že k všadeprítomnému narušeniu tvorby proteínovej štruktúry superoxiddismutázy 1 (SOD1) dochádza rovnakým spôsobom ako v priónoch. Predpokladá sa tiež, že inkorporácia β-metylamino-l-alanínu (BMAA) povedie k ďalšiemu priónovému šíreniu neusporiadanej tvorby proteínovej štruktúry. Ďalším bežným faktorom spojeným s ALS je lézia motorický systém v oblastiach, ako sú frontotemporálne laloky. Porážka v tejto oblasti je znakom skoré porušenia, ktoré možno použiť na predpovedanie straty motorickú funkciu. Mechanizmy ALS sa objavujú dlho predtým, ako sa objavia prvé príznaky a symptómy. Predtým, ako sa prejaví svalová atrofia, musí zomrieť asi jedna tretina motorických neurónov. Skúmalo sa mnoho ďalších potenciálnych rizikových faktorov – vystavenie chemikáliám, elektromagnetickým poliam, fyzickému zraneniu a úrazu elektrickým prúdom – ale neboli vyvodené žiadne konzistentné závery.
Patofyziológia
Charakteristickým rysom ALS je smrť horných a dolných motorických neurónov v projekčnej zóne mozgovej kôry, mozgového kmeňa a miechy. Pred ich smrťou sa v motorických neurónoch vyvinú inklúzie bohaté na proteíny v bunkovom tele a axónoch. Čiastočne to môže byť spôsobené degradáciou proteínov. Tieto inklúzie často obsahujú ubikvitín a často zahŕňajú jeden z ALS proteínov: SOD1, TAR DNA viažuci proteín (TDP-43 alebo TARDBP) alebo FUS RNA viažuci proteín.
Kostrové motorické jednotky
Vonkajšie svaly očnej gule a kostrové svaly vykazujú rôzne vlastnosti. Nižšie sú uvedené charakteristiky, ktoré odlišujú svaly očnej gule od kostrových svalov.
Jedno nervové vlákno sa spája iba s jedným svalovým vláknom
Napriek tomu žiadny strečový reflex veľké množstvo svalové vretená
Žiadna cyklická inhibícia
Nedostatok rýchlych/pomalých svalových zášklbov
Všetky motorické neuróny oka sa podieľajú na všetkých typoch pohybu očí.
Rozdiely sa pozorujú aj medzi zdravými a postihnutými svalmi očnej gule. Svaly očnej gule zosnulých darcov si zachovávajú svoju cytoarchitektoniku v porovnaní so svalmi končatín. Zdravé svaly očnej gule pozostávajú z centrálnej vrstvy (GL) pred očnou guľou a tenkej orbitálnej vrstvy (OL) pred očnicou. Okulomotorické svaly postihnuté ALS si zachovávajú polohu GL a OL. Okulomotorické svaly si zachovávajú neurotrofický faktor odvodený od mozgu (BDNF) a gliový neurotrofický faktor, ktoré sú tiež konzervované vo svaloch postihnutých ALS. Laminín je štrukturálny proteín lokalizovaný v nervovosvalovom spojení (NMJ). Lna4 je izoforma laminínu, ktorá je punc neuromuskulárne spojenie okohybných svalov. Ľudia s ALS si zachovávajú expresiu Lna4 na neuromuskulárnom okulomotorickom spojení, ale táto expresia chýba vo svaloch končatín tých istých ľudí. Udržiavanie expresie laminínu môže hrať dôležitú úlohu pri udržiavaní integrity okulomotorických svalov u ľudí s ALS. Ľudia so sporadickou ALS (sALS) majú zvýšená hladina intracelulárneho vápnika, ktorý spôsobuje zvýšené uvoľňovanie neurotransmiterov. Pasívny transport séra od ľudí so sALS zvyšuje spontánne uvoľňovanie mediátorov v cerebrospinálnej tekutine. Okulomotorické svaly sú odolné voči zmenám fyziologického stavu. Existuje však rôzne druhy vplyv choroby. Okulomotorické svaly postihnuté ALS majú väčšie rozdiely vo veľkosti vlákien ako zdravé kontrolné svaly. V okohybných svaloch boli nájdené zhluky a roztrúsené atrofické a hypertrofické vlákna, ale poškodenie týchto svalov je výrazne nižšie ako u svalov končatín tých istých darcov. Taktiež dochádza k nárastu spojivového tkaniva a nárastu tukového tkaniva v okohybných svaloch, aby sa kompenzovala strata vlákniny a atrofia. Pacienti s ALS majú tiež oftalmoplégiu, stratu neurónov okolo a v jadrách motorických svalov očnej gule. Okrem toho sa mení obsah ťažkého reťazca myozínu vo vláknach okohybných svalov, je narušená normálna expresia pomalého ťažkého reťazca myozínu v GL a v OL chýba embryonálny ťažký reťazec myozínu. Zmeny v ťažkom reťazci pomalého myozínu a v ťažkom reťazci embryonálneho myozínu sú jediné zmeny v okulomotorických svaloch. Keďže okulomotorický sval je vysoko inervovaný, akákoľvek denervácia je kompenzovaná susednými asconmi, ktoré zostávajú funkčné.
laktát a škoricovník
Kyselina mliečna - finálny produkt glykolýza, ktorá spôsobuje svalovú únavu. Enzým laktátdehydrogenáza pôsobí obojsmerne a dokáže oxidovať laktát na pyruvát, takže môže byť použitý v Krebsovom cykle. V okohybných svaloch laktát podporuje svalovú kontrakciu pri zvýšenej aktivite. Predpokladá sa, že okulomotorické svaly vysoká aktivita laktátdehydrogenázy sú odolné voči ALS. Cinnamate je blokátor transportu laktátu. Cinnamate je schopný spôsobiť únavu glozomotorických svalov, znižuje svalovú vytrvalosť a zvyškovú námahu. Cinnamát však nemá žiadny vplyv na dlhé svaly extenzorov na nohe. Nahradenie glukózy exogénnym laktátom zvyšuje únavu svalov extensor digitorum longus, ale nemá žiadny vplyv na glosomotorický sval. Únava okulomotorických svalov sa pozoruje iba vtedy, keď kombinácia exogénneho laktátu a cinamátu nahradí glukózu.
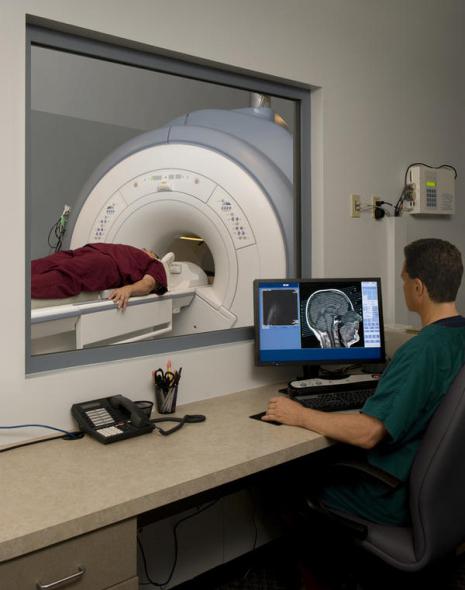
Diagnostika
Žiadna analýza nemôže presne diagnostikovať ALS, hoci prítomnosť znakov, ktoré naznačujú smrť horných a dolných motorických neurónov, je základným znakom. Diagnóza ALS sa vyskytuje predovšetkým na základe pozorovaní lekára a série testov, ktoré vylučujú iné ochorenia. Lekár odoberie kompletnú anamnézu pacienta a pravidelne vykonáva neurologické vyšetrenia na posúdenie progresie symptómov, ako je slabosť, svalová atrofia, hyperreflexia a svalová spasticita. Môžu sa vykonať ďalšie testy, aby sa vylúčila možnosť iných stavov, pretože mnohé zo symptómov ALS sa môžu vyskytnúť aj pri iných stavoch, ktoré možno liečiť. Jedným z takýchto testov je elektromyografia (EMG), ktorá zisťuje elektrickú aktivitu vo svaloch. Niektoré EMG nálezy môžu podporiť diagnózu ALS. Ďalším bežným testom je test rýchlosti nervového vedenia (NVR). Určitá odchýlka zistená v dôsledku testu môže naznačovať, že pacient má formu periférnej neuropatie (poškodenie periférne nervy) alebo myopatia (ochorenie svalov), nie ALS. Zobrazovanie magnetickou rezonanciou dokáže odhaliť abnormality, ktoré spôsobujú príznaky ALS, ako je opuch miecha, roztrúsená skleróza, herniovaný krčný disk, syringomyélia alebo cervikálna spondylóza. Na základe symptómov a výsledkov testov môže váš lekár nariadiť testy krvi a moču alebo iné laboratórne testy na vylúčenie iných možné choroby. V niektorých prípadoch, ak má lekár podozrenie, že pacient má skôr myopatiu ako ALS, môže tiež nariadiť svalovú biopsiu. Vírusové ochorenia, ako je vírus ľudskej imunodeficiencie (HIV), vírus T-bunková leukémiačlovek (HTLS), lymská borelióza, syfilis a kliešťová encefalitída môže spôsobiť príznaky podobné príznakom ALS. Neurologické ochorenia ako je roztrúsená skleróza, post-polio syndróm, multifokálna motorická neuropatia, Guillain-Barrého syndróm a spinálna svalová atrofia môžu mať tiež symptómy podobné ALS. Je dôležité správne diagnostikovať ochorenie, pretože príznaky ALS sa dajú ľahko zameniť s príznakmi mnohých iných chorôb. Preto je potrebné vyšetrenie kvalifikovaným neurológom, aby sa vylúčili iné ochorenia. Vo väčšine prípadov je ALS ľahko diagnostikovaná, pričom nesprávna diagnóza predstavuje menej ako 10 % prípadov. Uskutočnila sa štúdia s protokolom štúdie a pravidelnými vyšetreniami, ktorej sa zúčastnilo 190 pacientov, ktorí spĺňali kritériá pre MND/ALS. Diagnóza 30 pacientov (16 %) sa počas klinického sledovania dramaticky zmenila. V tej istej štúdii mali traja pacienti falošne negatívnu diagnózu, myasthenia gravis (TM, autoimunitné ochorenie). TM má rovnaké symptómy ako ALS a niekoľko ďalších neurologických abnormalít, čo vedie k oneskoreniu diagnózy a liečby. TM je liečiteľná, ale ALS nie. Myastenický syndróm, inak Lambertov-Eatonov syndróm, môže napodobňovať ALS a skoré príznaky podobné symptómom TM.
Liečba
U pacientov s ALS je potrebná terapia na zmiernenie symptómov a zvýšenie priemernej dĺžky života. Multidisciplinárny lekársky tím, ktorý pracuje s pacientmi s ALS, verí, že podporná starostlivosť je pre pacientov nevyhnutná, aby zostali aktívni a cítili sa pohodlne.
Lekárske prípravky
Riluzol (Rilutek) - mierne zvyšuje dĺžku života pacientov. Zvyšuje dĺžku života o niekoľko mesiacov a má väčší vplyv na pacientov s bulbárnou ALS. Droga vám tiež umožňuje oddialiť použitie ventilácie. Pacienti, ktorí užívajú liek, sa musia podrobiť vyšetreniu pečene (10 % ľudí má pri užívaní poškodenie pečene). Liek je schválený Ministerstvom zdravotníctva USA a odporúča sa ho používať. Národný inštitút klinická kvalifikácia. Riluzol nenapravuje už spôsobené poškodenie motorických neurónov. Iné lieky sa môžu použiť na zníženie únavy, zmiernenie svalových kŕčov, kontrolu spasticity a zníženie zvýšené slinenie a spúta. Množstvo liekov môže tiež zmierniť bolesť, depresiu, zlepšiť spánok, znížiť dysfágiu a zápchu. Baklofén a diazepam sú predpísané na kontrolu spasticity ALS. Ak majú pacienti problémy s prehĺtaním slín, môže sa predpísať trihexyfenidyl alebo amitriptylín.
Podpora dýchania
Keď sú svaly zapojené do dýchania oslabené, na podporu dýchania možno použiť ventiláciu (pretlaková ventilácia, dvojúrovňový pozitívny tlak v dýchacích cestách (BiPAP) alebo IVL bifázická ventilácia (BCV)). Takéto zariadenia umelo nútia fungovať pľúca pomocou vonkajších zariadení umiestnených na tvári a tele. Keď svaly už nedokážu udržať hladinu kyslíka a oxid uhličitý, tieto zariadenia je možné používať trvalo. BCV má výraznú výhodu v tom, že zariadenie dokáže vyčistiť sekréty pomocou vysokofrekvenčných vibrácií, ktoré sa vyskytujú po niekoľkých výdychoch. Pacienti môžu tiež zvážiť použitie mechanickej ventilácie (respirátorov), pri ktorej zariadenie nafúkne a vyfúkne pľúca. Pre efektívne využitie je potrebná hadička, ktorá musí prejsť z nosa alebo úst cez priedušnicu. Dlhodobé používanie takéhoto zariadenia si môže vyžadovať operáciu, tracheotómiu, počas ktorej sa priamo do priedušnice človeka zavedie cez otvor v krku. Pacienti a ich rodinní príslušníci by mali pri rozhodovaní, kedy použiť niektorý z vyššie popísaných zariadení a či ho vôbec použiť, zvážiť niekoľko faktorov. Ventilátory sa líšia svojim vplyvom na kvalitu života a cenou. Zatiaľ čo ventilácia môže pomôcť zmierniť problémy s dýchaním a tým predĺžiť život, neovplyvňuje progresiu ALS. Pred rozhodnutím, ktoré zariadenie si vybrať, by mali byť pacienti o nich a ich vplyve na život bez pohybu plne informovaní. Niektorí ľudia s predĺženou tracheotómiou, pretlakovou ventiláciou so špeciálnymi strojmi alebo hadicami môžu rozprávať, ak nie sú postihnuté svaly ústna dutina(ale v každom prípade s progresiou ochorenia dôjde k strate reči). Iní pacienti môžu používať hovoriaci ventil (ako napríklad Passy-Mure hovoriaci ventil) pod vedením logopéda. Externé ventilátory, ktoré pracujú v režime ventilácie BiPAP, sa používajú na udržanie dýchania najskôr v noci a potom cez deň. Používanie BiPAP je dočasné opatrenie. Dávno predtým, ako BiPAP prestane byť účinný, by pacienti mali zvážiť tracheotómiu a dlhodobú mechanickú ventiláciu. V tomto štádiu sa niektorí pacienti rozhodnú pre hospicovú liečbu.
Terapia
Fyzioterapia zohráva dôležitú úlohu v rehabilitačnom procese, priaznivo pôsobí na pacientov s ALS, umožňuje oddialiť stratu sily, udržať vytrvalosť, znížiť bolesť, predchádzať komplikáciám a zabezpečiť funkčnú nezávislosť. Rehabilitačná terapia a špeciálne vybavenie tiež pomáhajú zabezpečiť nezávislosť a bezpečnosť pacientov v priebehu ALS. Ľahké aeróbne cvičenie, ako je chôdza, plávanie a bicyklovanie, posilňuje nepostihnuté svaly, zlepšuje kardiovaskulárne zdravie a pomáha pacientom vyrovnať sa s únavou a depresiou. Striedavý pohyb a naťahovacie cvičenia bránia svalovým kŕčom a sťahovaniu svalov. Terapeuti musia zabezpečiť, aby boli svaly zaťažené bez toho, aby sa prepracovali. Rampy, výstuhy, chodítka, kúpeľňové vybavenie, invalidné vozíkyčo pomáha pacientom zostať mobilnými. Terapeuti môžu odporučiť vybavenie alebo zariadenia, ktoré pomôžu pacientom zostať v bezpečí a čo najnormálnejší. Pacienti, ktorí majú problémy s rozprávaním, môžu spolupracovať so špecialistami na poruchy reči, aby pomohli pacientom naučiť techniky, ako napríklad hovoriť hlasnejšie a jasnejšie. S progresiou ochorenia môžu odborníci odporučiť používanie pomôcok na zlepšenie reči, príp alternatívne spôsoby komunikácia, ako sú reproduktory, zariadenia generujúce reč a/alebo abecedné tabule, komunikácia prostredníctvom signálov áno/nie.
Výživa
Pacienti a ich opatrovatelia môžu dostávať potrebné informácie od odborníkov na výživu o tom, ako si naplánovať svoj jedálniček a jesť malé jedlá počas dňa, ktoré poskytnú dostatok kalórií, vlákniny a tekutín, ako aj informácie o tom, akým potravinám sa vyhnúť, aby ste sa vyhli problémom s prehĺtaním. Pacienti môžu použiť odsávacie zariadenia na odstránenie prebytočnej tekutiny a slín, čím zabránia uduseniu. Terapeuti môžu pomôcť s odporúčaniami pre samokŕmenie. Špecialisti na poruchy reči vám môžu pomôcť vybrať produkty, ktoré najlepšie vyhovujú vašim schopnostiam. Keď pacient už nemôže prijímať živiny z potravy môžu lekári odporučiť použitie sondy na kŕmenie. Používanie kŕmnej trubice tiež zabraňuje riziku udusenia a zápalu pľúc, ktoré môže vyplynúť z vdýchnutia tekutiny do pľúc. Slúchadlo nespôsobuje žiadne bolesť a umožňuje pacientom jesť sami, ak si to želajú. Vedci uvádzajú, že „pacienti s ALS majú chronický deficit príjmu energie“ a zníženú chuť do jedla. Štúdie na zvieratách a ľuďoch potvrdzujú, že pacienti by mali konzumovať čo najviac kalórií a nikdy by nemali znižovať príjem kalórií. Od roku 2012 neexistujú presné údaje týkajúce sa odtučňovacej liečby.
Paliatívnej starostlivosti
Sociálni pracovníci, opatrovatelia a hospicové sestry pomáhajú pacientom s ALS, ich rodinám a opatrovateľom vyrovnať sa so zdravotnými, emocionálnymi a finančnými ťažkosťami, najmä v pokročilých štádiách ochorenia. Sociálni pracovníci poskytujú podporu pri získaní finančnej pomoci, spísaní plnej moci a závetu a pomáhajú nájsť podporu pre rodiny a opatrovateľov. Opatrovatelia nielen poskytujú zdravotná starostlivosť, ale aj naučiť rodinných príslušníkov pacienta, ako správne používať respirátory, kŕmiť a presúvať pacienta tak, aby sa predišlo bolestivým kožným problémom a napätiu. Hospicové sestry spolupracujú s lekármi pri poskytovaní vhodnej starostlivosti a pomoci pri iných otázkach súvisiacich so zlepšovaním kvality života pacientov, ktorí chcú zostať doma. Pracovníci hospicu tiež radia pacientom a ich rodinám vo všetkých otázkach súvisiacich s konečným štádiom ochorenia.
Epidemiológia
Vo väčšine krajín nie je výskyt ALS známy. V Európe toto ochorenie postihuje približne 2,2 ľudí na 100 000 ľudí ročne. V USA je každý rok diagnostikovaných 5 600 ľudí, zatiaľ čo 30 000 Američanov už má túto chorobu. Zo strany amyotrofická skleróza každý rok zomierajú dvaja zo 100 000 ľudí. Zvažuje sa ALS zriedkavé ochorenie, ale najčastejšie zo všetkých ochorení motorických neurónov, ktoré postihuje ľudí akejkoľvek rasy a etnického pôvodu. ALS sa ročne vyvinie u 1-2 ľudí na 100 000. ALS postihuje až 30 000 Američanov. Uvádza sa, že toto ochorenie postihuje 1,2–4,0 na každých 100 000 belochov, pričom menšie počty sa pozorujú v iných etnických skupinách. Druhú najvyššiu prevalenciu tohto ochorenia majú Filipínci (1,1 – 2,8 na každých 100 000). Existujú správy o niekoľkých „skupinách“ vrátane troch hráčov amerického futbalu zo San Francisco 49ers, viac ako 50 futbalových hráčov z Talianska, troch futbalových priateľov na juhu Anglicka a rodinných prípadov (manžela a manželky) na juhu Francúzska. Väčšina výskumníkov tvrdí, že ALS sa vyskytuje v dôsledku kombinácie dedičných a enviromentálne faktory, hoci to druhé nebolo potvrdené, na rozdiel od zvýšeného rizika ochorenia s vekom.
Príbeh
Chorobu prvýkrát opísal v roku 1824 Charles Bell. V Spojených štátoch sa choroba stala známou po tom, čo zasiahla slávneho bejzbalového hráča Lou Gehriga, a potom po kampani s názvom ice bucket challenge v roku 2014. Anglický vedec August Waller opísal vzhľad nervové vlákna v roku 1850. V roku 1869 spojenie medzi symptómami a základnými neurologickými problémami opísal Jean-Martin Charcot, ktorý vo svojej práci v roku 1874 zaviedol koncept amyotrofickej laterálnej sklerózy. V roku 1881 bol článok preložený do anglický jazyk a publikované v troch zväzkoch Prednášok o chorobách nervový systém". ALS sa stala známou v Spojených štátoch v roku 1939 po tom, čo choroba postihla baseballovú legendu Lou Gehriga, ktorý zomrel o dva roky neskôr. V 50-tych rokoch sa medzi obyvateľmi Chamorro na Guame vyskytla epidémia ALS. V roku 1991 už vedci spájali chromozóm 21 s dedičnou formou ALS (HALS). V roku 1993 sa zistilo, že gén SOD1 na chromozóme 21 hrá dôležitú úlohu v mnohých prípadoch dedičnej formy ochorenia. V roku 1996 bol riluzol schválený Ministerstvom zdravotníctva USA na liečbu ALS. V roku 1998 bolo ustanovené kritérium El Escorial ako štandard na klasifikáciu pacientov s ALS v klinických štúdiách. Nasledujúci rok bola zverejnená škála funkčnosti ALS, ktorá sa stala štandardom na hodnotenie ochorenia v klinických štúdiách. V roku 2011 sa zistilo, že viacnásobné opakovania C9ORF72 sú hlavnou príčinou ALS a frontotemporálnej demencie.
Etymológia
Výraz „amyotrofný“ pochádza z gréckeho slova amyotrofia: a- znamená „nie“, myo znamená „sval“ a trofia znamená „výživa“; teda amyotrofia znamená „nedostatok výživy svalov“, čo presne vystihuje charakteristický znak ochorenie, svalová atrofia. "Laterálny" označuje oblasť ľudskej miechy, kde sa nachádzajú postihnuté nervové bunky. Degenerácia v tejto oblasti vedie k zhutneniu, skleróze.
Verejná podpora a kultúrny odkaz
V auguste 2014 sa na internete uskutočnila akcia na podporu ľudí s ALS s názvom ALS Ice Bucket Challenge. Demonštrant musel naplniť vedro vody ľadom, potom pomenovať osobu, ktorá ho vyzvala, a tiež vymenovať troch ľudí, ktorých vyzval. Potom na seba účastník zvalil vedro s vodou a ľadom. Do akcie sa ale dalo zapojiť aj inak. Člen môže darovať minimálne 10 USD (alebo ekvivalent v inej mene) na výskum ALS v Spojenom kráľovstve. Ten, kto nechce zmoknúť studená voda musí venovať minimálne 100 $ na výskum ALS. K 25. augustu akcia vyzbierala 79,7 milióna dolárov, zatiaľ čo v roku 2013 sa vyzbieralo iba 2,5 milióna dolárov.Na tejto akcii sa zúčastnilo mnoho celebrít. ALS je stredobodom filmu You Are Not You z roku 2014, v ktorom hrajú Hilary Swank, Emily Rossum a Josh Duhamel.
Výskum
Klinické skúšky prebiehajú po celom svete; zoznam klinických štúdií v USA možno nájsť na stránke ClinicalTrials.gov. Najväčšia genetická štúdia s názvom projekt MinE stále prebieha. Projekt je financovaný z verejných zdrojov a zahŕňa mnoho krajín. Fáza II štúdie bola dokončená a fáza IIb stále prebieha pod názvom „BENEFIT-ALS“. Výsledky prvej štúdie sú k dispozícii tu Súčasná štúdia je medzinárodná, placebom kontrolovaná štúdia zahŕňajúca 680 pacientov. To z nej robí doteraz najväčšiu štúdiu. V súčasnosti prebieha štúdia fázy II o protilátke ozanezumab. Ide o veľkú štúdiu sponzorovanú britskou spoločnosťou GlaxoSmithKline. Nemocnica Hadaas v Izraeli v štádiu II klinická štúdia vedená spoločnosťou BrainStorm Cell Therapeutics, práca je zameraná na stabilizáciu parametrov funkčnej škály ALS. Kmeňové bunky sa počas testu odstránia kostná dreňčloveka a diferencujú sa do voľného priestoru bunky, čím sa aktivujú neurotropné faktory. Bunky sa injikujú späť tomu istému pacientovi prostredníctvom intratekálnych a intramuskulárnych injekcií. Plánuje sa, že druhá časť fázy II sa uskutoční v niekoľkých amerických inštitúciách vrátane Mayo Clinic.
Ochorenie amyotrofická laterálna skleróza (ALS) sa vyskytuje u troch zo stotisíc ľudí. Napriek dnešnému pokroku v medicíne je úmrtnosť na túto patológiu 100%. Sú však prípady, keď pacienti po čase nezomreli, ich stav sa stabilizoval. Vzorový príklad je známy gitarista Jason Becker. S touto chorobou aktívne bojuje už viac ako 20 rokov.
čo je BAS?
Pri tejto chorobe dochádza k neustálej smrti motorických neurónov miechy a jednotlivých častí mozgu, ktoré sú zodpovedné za dobrovoľné pohyby. Časom svaly u ľudí s touto diagnózou atrofujú, keďže sú neustále neaktívne. Ochorenie sa prejavuje vo forme ochrnutia končatín, svalov tela a tváre.
Amyotrofická laterálna skleróza sa nazýva amyotrofická laterálna skleróza iba preto, že neuróny, ktoré vedú impulzy do všetkých svalov, sú umiestnené na oboch stranách miechy. Posledná fáza ochorenia je diagnostikovaná, keď sa patologický proces dostane do dýchacieho traktu. Smrť nastáva v dôsledku svalovej atrofie alebo infekcie. V niektorých prípadoch sú dýchacie svaly postihnuté skôr ako končatiny. Človek zomiera veľmi rýchlo bez toho, aby zažil všetky útrapy života s paralýzou.
V mnohých európskych krajinách je amyotrofická laterálna skleróza známa ako Lou Gehrigova choroba. Tento slávny hráč baseballu z Ameriky bol diagnostikovaný už v roku 1939. Len za pár rokov úplne stratil kontrolu nad svojím telom, jeho svaly boli vyčerpané a samotný športovec sa stal invalidom. Lou Gehrig zomrel v roku 1941.
Rizikové faktory
V roku 1865 Charcot (francúzsky neurológ) prvýkrát opísal túto chorobu. Dnes ňou na celom svete netrpí viac ako päť ľudí zo stotisíc. Vek pacientov s touto diagnózou sa pohybuje od 20 do 80 rokov. Zástupcovia silnejšieho pohlavia sú náchylnejší na túto chorobu.
V 10% prípadov je ALS ochorenie zdedené. Vedci identifikovali asi 15 génov, ktorých mutácia v rôznej miere sa prejavuje u ľudí s touto patológiou. 
Zvyšných 90 % prípadov má sporadický charakter, to znamená, že nesúvisí s dedičnosťou. Odborníci nevedia menovať konkrétne dôvodyčo vedie k rozvoju ochorenia. Predpokladá sa, že niektoré faktory môžu stále zvyšovať riziko ochorenia, a to:
- Fajčenie.
- Práca v nebezpečnom priemysle.
- Služba v armáde (vedci tento jav ťažko vysvetľujú).
- Jesť potraviny, ktoré boli vypestované pesticídmi.
Hlavné príčiny ochorenia
Závažný patologický proces sa môže začať úplne rôznych faktorov s ktorými sa denne stretávame skutočný život. Prečo vzniká ALS? Dôvody môžu byť nasledovné:
- Intoxikácia tela ťažkými kovmi.
- Infekčné choroby.
- Nedostatok niektorých vitamínov.
- Poranenie elektrickým prúdom.
- Tehotenstvo.
- Zhubné novotvary.
- Chirurgické zákroky (odstránenie časti žalúdka).
Formy ochorenia
Cervikotorakálna forma je charakterizovaná rozšírením patologického procesu do oblasti lopatiek, paží a všetkého ramenného pletenca. Pre človeka je postupne ťažké vykonávať zaužívané pohyby (napríklad zapínanie gombíkov), na ktoré sa pred chorobou nemusel sústrediť. Keď ruky prestanú "poslúchať", dôjde k úplnej svalovej atrofii. 
Lumbosakrálna forma je charakterizovaná porážkou dolných končatín ako ruky. Postupne sa rozvíja slabosť svalov v tejto oblasti, objavujú sa zášklby a kŕče. Pacienti začínajú pociťovať ťažkosti s chôdzou, neustále sa potkýnajú.
Bulbárna forma je jedným z najzávažnejších prejavov ochorenia. Pacientom sa veľmi zriedka podarí prežiť viac ako štyri roky od nástupu primárnych symptómov. Príznaky ochorenia ALS začínajú problémami s rečou a nekontrolovateľnou mimikou. Pacienti majú ťažkosti s prehĺtaním, ktoré sa mení na úplnú neschopnosť samostatne jesť. Patologický proces, zachytávajúci celé ľudské telo, negatívne ovplyvňuje prácu dýchacích a kardiovaskulárnych systémov. Preto pacienti s touto formou zomierajú skôr, ako sa rozvinie paralýza.
Mozgová forma je charakterizovaná súčasným zapojením do patologického procesu horných aj dolných končatín. Okrem toho môžu pacienti bez dôvodu plakať alebo sa smiať. Podľa závažnosti cerebrálna forma nie je nižšia ako bulbárna, takže smrť z nej prichádza rovnako rýchlo.
Klinický obraz
Podľa niektorých údajov dokonca aj v predklinickom štádiu odumiera približne 80 % motorických neurónov. Všetku ich prácu preberajú susedné bunky. Postupne zvyšujú počet koncových vetiev a takzvaný iónový kód sa začína prekladať na veľké množstvo svalov. V dôsledku vytvoreného preťaženia odumierajú aj tieto neuróny. Takto začína amyotrofická laterálna skleróza. Symptómy ochorenia sa neprejavia bezprostredne po smrti motorických neurónov.
Môže trvať 5-7 mesiacov, kým človek začne venovať pozornosť vonkajšie zmeny svojho tela. Pacienti majú spravidla pokles telesnej hmotnosti a svalovú slabosť, sú ťažkosti s vykonávaním každodenných činností. Je ťažké normálne sa pohybovať, nosiť predmety v rukách, dýchať, prehĺtať a hovoriť. Objavujú sa kŕče a zášklby. Takéto príznaky sú charakteristické pre mnohé ochorenia, čo výrazne komplikuje diagnostiku ALS v počiatočných štádiách vývoja.
S touto patológiou nie sú systémy nikdy ovplyvnené vnútorné orgány(obličky, pečeň, srdce), svaly zodpovedné za črevnú motilitu.
Ochorenie ALS má progresívny charakter a postupom času postihuje čoraz viac oblastí tela. Osoba postupne stráca schopnosť pohybovať sa bez ťažkostí v dôsledku porušenia prehĺtacie reflexy jedlo je neustále v Dýchacie cestyčo spôsobuje prerušenia dýchania. V posledných fázach je vitálna aktivita podporovaná len vďaka umelej výžive a ventilátoru. 
Diagnostika
Diagnóza ochorenia je mimoriadne náročná. Celá pointa je v tom počiatočné štádiá Ochorenie ALS má podobné črty ako iné neurologické poruchy. Až po dôkladnom vyšetrení pacienta môže lekár stanoviť konečnú diagnózu.
Diagnóza zahŕňa mnohostranné štúdium zdravotného stavu pacienta, počnúc zberom anamnézy a končiac molekulárno-genetickou analýzou. Okrem toho v celkom určite neurologické vyšetrenie, MRI, sérologické a biochemické analýzy krvi. 
Aká by mala byť liečba?
V súčasnosti odborníci nevedia ponúknuť efektívne metódy liečbe. Všetka pomoc lekárov sa obmedzuje na maximálne uľahčenie prejavov choroby.
Liečba amyotrofickej laterálnej sklerózy zahŕňa užívanie anticholínesterázových liekov (Galantamín, Prozerin) na zlepšenie kvality reči a prehĺtania, myorelaxancií (Diazepam), antidepresíva a trankvilizéry. Pri infekčných léziách je predpísaná antibiotická terapia. Kedy silná bolesť lekári odporúčajú užívať nesteroidné protizápalové lieky, následne sa nahrádzajú omamnými látkami.
jediný účinný liekúčelová akcia je Rilutek. Nielenže predlžuje život pacienta, ale tiež umožňuje predĺžiť oneskorenie pri prechode na ventilátor. 
Dobrá starostlivosť výrazne zlepšuje kvalitu života
Samozrejme, každý človek, ktorému bola diagnostikovaná ALS, potrebuje náležitá starostlivosť. Pacient kriticky skúma svoj stav, pretože každý deň jeho telo doslova bledne. Nakoniec sa takíto ľudia prestanú samostatne obsluhovať, komunikovať s príbuznými a priateľmi a upadajú do depresie.
Všetci pacienti s ALS bez výnimky potrebujú:
- Vo funkčnej posteli so špeciálnym zdvíhacím mechanizmom.
- V záchodovej doske.
- Na invalidnom vozíku s automatickým ovládaním.
- V komunikačných prostriedkoch napríklad v notebooku.
Zvláštnu pozornosť si vyžaduje aj výživa pacientov. Je lepšie podávať dobre prehltnuté jedlo bohaté na vitamíny a bielkoviny. Následne výživa bez pomoci špeciálnej sondy nie je možná.
Amyotrofická laterálna skleróza sa u niektorých ľudí rýchlo rozvíja. Príbuzní a priatelia to majú veľmi ťažké, pretože človek nám doslova mizne pred očami. Ľudia, ktorí sa starajú o chorých, často potrebujú ďalšiu pomoc od psychológa, ako aj užívanie sedatív.
Predpoveď
Ak lekár potvrdil amyotrofickú laterálnu sklerózu, príznaky zo dňa na deň len pribúdajú, zhoršujú sa všeobecný stav pacient, prognóza je v tomto prípade sklamaním. V celej histórii modernej medicíny boli zaznamenané len dva prípady, kedy sa pacientom podarilo prežiť. O prvom sme už hovorili v tomto článku. A druhým je slávny fyzik Stephen Hawking, ktorý s takýmto ochorením naďalej úspešne existuje posledných 50 rokov svojho života. Vedec aktívne pracuje a teší sa z každého nového dňa, hoci sa pohybuje na špeciálne vybavenom kresle a s ostatnými komunikuje prostredníctvom počítačového syntetizátora reči. 
Preventívne opatrenia
O primárna prevencia nie je potrebné hovoriť o patológii, pretože presné dôvody jej výskytu stále nie sú jasné. Sekundárna prevencia by mala byť zameraná na spomalenie progresie ochorenia a predĺženie života pacienta. Obsahuje:
- Pravidelné konzultácie s neurológom a užívanie liekov.
- Úplné odmietnutie všetkých zlé návyky pretože len zhoršujú ALS chorobu.
- Liečba by mala byť adekvátna a kompetentná.
- Vyvážená a racionálna výživa.
Záver
V tomto článku sme hovorili o tom, čo predstavuje ochorenie ALS. Príznaky a liečba tohto patologický stav netreba ignorovať. Bohužiaľ, moderná medicína nemôže ponúknuť účinnú terapiu proti tejto chorobe. Príbuzní a priatelia by však mali vynaložiť maximálne úsilie na zlepšenie každodenný život osoba s touto diagnózou.
Amyotrofickú laterálnu sklerózu (ALS, "Hehrigova choroba", "ochorenie motorických neurónov") prvýkrát opísal francúzsky psychiater Martin Charcot v roku 1869.
V USA a Kanade existuje ďalší pojem – „Lou Gehrigova choroba“, známy hráč bejzbalu, ktorý musel vo veku 36 rokov ukončiť kariéru pre amyotrofickú laterálnu sklerózu.
Čo je amyotrofická laterálna skleróza?
Amyotrofická laterálna skleróza je ochorenie nervového systému, ktoré rýchlo postihuje motorické neuróny miechy, mozgového kmeňa a kôry.
IN patologický proces zúčastniť sa motorické nervy kraniálne neuróny (tvárové, ternárne, glosofaryngeálne).
Amyotrofická laterálna skleróza je zriedkavá (2-3 ľudia na 100 000) a rýchlo postupuje.
V medicíne existuje ďalší koncept - syndróm amyotrofickej laterálnej sklerózy. Je vyvolaná inou chorobou, takže liečba v tomto prípade je zameraná na odstránenie základnej patológie. Ak má pacient príznaky ALS, ale ich príčiny nie sú známe, lekári nehovoria o syndróme, ale o chorobe.
Pri ALS sú motorické neuróny zničené, prestávajú vysielať signály z mozgu do svalov, v dôsledku čoho svaly začínajú slabnúť a atrofovať.
Príčiny

Dôvody, ktoré vyvolávajú výskyt tohto ochorenia, ešte neboli stanovené. Vedci však ponúkajú niekoľko teórií:
dedičné
Zistilo sa, že v 10-15% prípadov je choroba dedičná.
Vírusové
Táto teória sa rozšírila v 60. rokoch 20. storočia v USA a ZSSR. V tom čase sa robili pokusy na opiciach. Zvieratám sa injekčne podávali extrakty z miechy chorých ľudí. Predpokladalo sa tiež, že ochorenie môže vyvolať vírus detskej obrny.
Gennaya
Narušenie génu sa zistí u 20 % pacientov s ALS. Kódujú enzým superoxid dismutáza-1, ktorý sa stáva nebezpečným pre nervové bunky superoxid na kyslík.
autoimunitné
Vedci vykonali výskum a našli protilátky, ktoré zabíjajú motorické nervové bunky. Je dokázané, že tieto protilátky sa môžu vytvárať, keď vážnych chorôb(Hodgkinov lymfóm, rakovina pľúc atď.).
neurálny
Túto teóriu vyvinuli britskí vedci, ktorí veria, že tvorba ALS môže vyvolať prvky glií - buniek zodpovedných za životne dôležitú aktivitu neurónov. Ak je narušená funkcia astrocytov, ktoré odstraňujú glutamát z nervových zakončení, riziko Charcotovej choroby sa niekoľkonásobne zvyšuje.
Medzi rizikové faktory lekári rozlišujú: dedičnú predispozíciu, vek nad 50 rokov, fajčenie, prácu spojenú s používaním olova, vojenskú službu.
Príznaky ALS
Bez ohľadu na formu ochorenia všetci pacienti pociťujú svalovú slabosť, objavujú sa svalové zášklby, svalová hmota ubúda.
Svalová slabosť sa rýchlo zvyšuje, ale očné svaly a zvierač močového mechúra nie sú ovplyvnené.
V počiatočnom štádiu ochorenia sa vyskytuje:
- svalová slabosť v členkoch a chodidlách;
- atrofia ruky;
- zhoršené motorické zručnosti a reč;
- ťažkosti s prehĺtaním;
- svalové zášklby;
- kŕče jazyka, rúk a ramien.
S rozvojom ALS sa objavujú záchvaty smiechu a plaču, narúša sa rovnováha, objavuje sa atrofia jazyka.
Kognitívne funkcie sa zhoršujú len v 1-2 % prípadov ochorenia, u ostatných pacientov sa duševná aktivita nemení.
V neskorších štádiách sa u pacienta rozvinie depresia, začnú sa prerušovať dýchanie, stráca sa schopnosť samostatného pohybu.
Pacienti s ALS sa prestávajú zaujímať o svojich blízkych a vonkajší svet, stávajú sa rozmarnými, neviazanými, emočne labilnými a agresívnymi. Keď dýchacie svaly prestanú pracovať, osoba potrebuje mechanickú ventiláciu.
Priebeh ochorenia
Spočiatku sa objavujú príznaky, ktoré bránia človeku viesť plnohodnotný život: svalová necitlivosť, kŕče, zášklby, ťažkosti s rozprávaním. Ale spravidla je na samom začiatku ťažké určiť presnú príčinu týchto porúch.
Vo väčšine prípadov sa ALS uvádza do štádia svalovej atrofie.
Postupne sa svalová slabosť šíri a pokrýva nové časti tela, pacient sa nemôže samostatne pohybovať, začínajú problémy s dýchaním.
Pacienti s ALS zriedka trpia demenciou, ale ich stav vedie k ťažkej depresii v očakávaní smrti. V poslednom štádiu už človek nemôže sám jesť, chodiť a dýchať, potrebuje špeciálne zdravotnícke pomôcky.
Formy ochorenia
Formy ochorenia sa vyznačujú lokalizáciou poškodených svalov.
Bulbarnaya
Postihnuté sú hlavové nervy (9,10,12 párov).
Pacienti s bulbárnou formou ALS začínajú mať problémy s rečou, sťažujú sa na ťažkosti s výslovnosťou, je pre nich ťažké hýbať jazykom.
S progresiou ochorenia je akt prehĺtania narušený, jedlo sa môže vyliať cez nos. V neskorom štádiu ochorenia svaly tváre a krku úplne atrofujú, mimika mimiky a pacienti s ALS nemôžu otvárať ústa a žuť jedlo.
cervikotorakálny

Ochorenie postupuje v horných končatinách na oboch stranách.
Spočiatku sa objavuje nepohodlie v rukách, pre človeka je ťažké vykonávať zložité pohyby rukami, písať, hrať na hudobné nástroje. Pri vyšetrení si lekár všimne, že svaly paží pacienta sú napäté a reflexy šliach sú zvýšené.
V pokročilých štádiách ochorenia svalová slabosť progreduje a šíri sa na predlaktia a ramená.
lumbosakrálny
![]()
Prvým príznakom je slabosť dolných končatín.
Pre pacienta je ťažšie vykonávať prácu v stoji, stúpať po schodoch, jazdiť na bicykli a chodiť na dlhé vzdialenosti.
V priebehu času začne noha ochabovať, chôdza sa zmení, potom svaly nôh úplne atrofujú, človek nemôže chodiť, vzniká inkontinencia moču a stolice.
Takmer 50 % pacientov trpí cervikotorakálnou formou ALS, 25 % je lumbosakrálnych a bulbárnych.
Diagnostika
Neurológ používa ako hlavné diagnostické metódy:
MRI mozgu a miechy
Pomocou tejto metódy je možné odhaliť degeneráciu pyramídových štruktúr a atrofiu motorických častí mozgu.
Neurofyziologické vyšetrenia
Na detekciu ALS sa používa TKMS (transkraniálna magnetická stimulácia), ENG (elektroneurografia), EMG (elektromyografia).
Cerebrospinálna punkcia
Stanoví sa hladina obsahu bielkovín (normálna alebo zvýšená).
Biochemické krvné testy
U pacientov s ALS sa zistilo päťnásobné alebo viacnásobné zvýšenie kreatínfosfokinázy, akumulácia kreatinínu a močoviny a zvýšenie AST a ALT.
Molekulárna genetická analýza
Gén kódujúci superoxiddismutázu-1 sa skúma.
Ale tieto metódy nestačia na identifikáciu diagnózy súčasne použité odlišná diagnóza potvrdiť alebo vylúčiť choroby:
- mozog: dyscirkulačná encefalopatia, nádory zadnej lebečnej jamky, multisystémová atrofia.
- miecha: nádory, syringomyelia, lymfocytová leukémia, spinálna amyotrofia atď.
- svaly: myozitída, okulofaryngeálna myodystrofia, myotónia Rossolimo-Steiner-Kurshman.
- periférne nervy: multifokálna motorická neuropatia, Parsonage-Turnerov syndróm atď.
- neuromuskulárnu synapsiu: Lambertov-Eatonov syndróm, myasténia gravis.
Liečba
Neexistuje žiadny liek na ALS, ale je možné spomaliť progresiu tejto choroby, zvýšiť dĺžku života a zmierniť stav človeka.
Na tento účel sa používa komplexná terapia:
Liek, ktorý bol prvýkrát použitý pri liečbe ALS vo Veľkej Británii a USA. Účinné látky blokujú uvoľňovanie glutamínu a spomaľujú proces poškodenia neurónov. Liek sa musí užívať 2-krát denne po 0,05 g.
Svalové relaxanciá a antibiotiká pomoc pri svalovej slabosti. Na odstránenie svalových kŕčov a zášklbov sú predpísané Mydocalm, Baclofen, Sirdalud.

Pre zvýšenie svalová hmota používa sa anabolický "Retabolin".
Antibiotiká ak sa rozvinie sepsa resp infekčné komplikácie. Lekári predpisujú fluorochinolóny, cefalosporíny, karbopény.
vitamíny skupiny B, E, A, C na zlepšenie impulzu pozdĺž nervových vlákien.
Anticholínesterázové lieky, ktoré spomaľujú proces deštrukcie acetylcholínu ("Kalimin", "Prozerin", "Pyridostigmín").

V niektorých prípadoch sa používa transplantácia kmeňových buniek. Zabraňuje odumieraniu nervových buniek, podporuje rast nervových vlákien a obnovuje nervové spojenia.
V neskorších fázach použite antidepresíva a trankvilizéry, nesteroidné lieky proti bolesti a opiáty.
Ak je spánok narušený, predpisujú sa benzodiazepínové prípravky.
Na uľahčenie pohybu stoličky a postele s rôzne funkcie, palice, fixačné obojky. Lekári odporúčajú logopédiu. V neskorších štádiách ochorenia bude potrebný odsávač slín a následne tracheostómia, aby pacient mohol dýchať.
Netradičné metódy liečby ALS nedávajú pozitívny výsledok.
Predpoveď a dôsledky
Prognóza pre pacientov s ALS nepriaznivé. Smrteľný výsledok nastáva o 2-12 rokov, ako ťažký zápal pľúc, respiračné zlyhanie alebo iné vážna choroba spôsobené Gehrigovou chorobou.
Pri bulbárnej forme a u starších pacientov sa obdobie skracuje na 3 roky.
Prevencia
Opatrenia na prevenciu lieku ALS sú stále neznáme.
Choroba postupuje veľmi rýchlo, neexistujú žiadne prípady úspešnej liečby a obnovy motorických funkcií tela. Svalová slabosť, ktorá sa postupne zvyšuje, úplne mení život človeka a jeho rodiny.
Ale napriek neuspokojivým prognózam a nedostatočnému štúdiu choroby by blízki mali dúfať, že v blízkej budúcnosti budú vyvinuté účinné liečby. terapeutické metódy liečbe. Medzitým je potrebné prijať opatrenia na zmiernenie stavu osoby s ALS.
amyotrofická laterálna skleróza - nebezpečná choroba ktoré môžu znehybniť osobu a viesť k smrti.
Lekári stále nedokážu určiť presné príčiny ochorenia a nájsť účinnú liečbu. V súčasnosti môže medicína iba zmierniť stav pacientov s ALS. Ani jeden pacient sa z tejto choroby nedokázal úplne vyliečiť. Je dôležité odlíšiť „chorobu ALS“ od „syndrómu ALS“. V druhom prípade je prognóza zotavenia oveľa lepšia.
(ALS), tiež známa ako Lou Gehrigova choroba, je pomaly progresívne, nevyliečiteľné degeneratívne ochorenie centrálneho nervového systému. Podľa Asociačného štátu len polovica obyvateľov USA počula o tejto chorobe, rovnaký vzorec sa pozoruje aj v iných krajinách.
Jeden spôsob, ako upútať pozornosť k problému amyotrofickej laterálnej sklerózy sa stala Ice Bucket Challenge, v ktorej sa ľudia musia obliať vedrom ľadovej vody a prispieť. V auguste 2014 si kampaň získala mimoriadnu popularitu po celom svete a podarilo sa jej prilákať 50 miliónov dolárov v podobe darov a viac ako 1,5 milióna účastníkov. prezident a CEO Spoločnosť 3 M Inge Thulin sa pridal k počtu účastníkov a vyjadril sa k svojej účasti na akcii:
"amyotrofická laterálna skleróza(BAS) — hrozná choroba. Prijal som výzvu od rodiny nášho zamestnanca 3M Allena Wahlgrena, ktorý týmto stavom trpí už vyše 32 rokov. Diagnostikovali mu ju začiatkom roka a dnes je už takmer úplne ochrnutý. Presne pred rokom sme prišli aj o jedného z najlepších lídrov v dentálnom biznise 3M Larryho Leera, ktorý zomrel z ALS. Videl som, ako rýchlo „vyhorel“, bolo to hrozné. A túto výzvu som prijal nielen na počesť Allena a Larryho, ale aj na počesť všetkých rodín, ktoré čelia tejto hroznej chorobe.“
Príčiny ochorenia ALS
Príčinou ALS je mutácia niektorých proteínov (ubikvitínu) s výskytom intracelulárnych agregátov. Rodinné formy ochorenia sa pozorujú v 5% prípadov. Ochorenie ALS v podstate postihuje ľudí nad štyridsaťšesťdesiat rokov, z ktorých nie viac ako 10 % je nositeľmi dedičnej formy, zvyšok prípadov vedci stále nevedia vysvetliť vplyvom akéhokoľvek vonkajšie vplyvy- ekológia, úrazy, choroby a iné faktory.
Príznaky ochorenia
Včasné príznaky choroby sú kŕče, zášklby, necitlivosť a slabosť v končatinách, ako aj ťažkosti s rečou, ale takéto príznaky sa týkajú veľkého počtu chorôb. To veľmi sťažuje diagnostiku až do záverečného obdobia, choroba už prechádza do štádia svalovej atrofie.
Počiatočné ALS lézie sa môžu vyskytnúť na rôzne časti tela, pričom až u 75 % pacientov sa ochorenie začína na končatinách, hlavne dolných.
Čo to je? Ako sa to prejavuje
Počiatočné prejavy choroby:
.slabosť v distálnych častiach rúk, nemotornosť pri vykonávaní jemných pohybov prstami, chudnutie na rukách a fascikulácie (svalové zášklby)
.menej často choroba začína slabosťou proximálnych ramien a ramenného pletenca, atrofiou svalov nôh v kombinácii s dolnou spastickou paraparézou
Je tiež možný nástup ochorenia s bulbárnymi poruchami - dyzartria a dysfágia (25% prípadov)
Kŕče (bolestivé kontrakcie, svalové kŕče), často generalizované, sa vyskytujú takmer u všetkých pacientov s ALS a sú často prvým príznakom ochorenia
charakteristický klinické prejavy BAS
Amyotrofická laterálna skleróza je charakterizovaná kombinovanou léziou dolného motorického neurónu (periférne) a léziou horného motorického neurónu (pytamidové dráhy a / alebo pyramídové bunky motorickej kôry mozgu.
Príznaky poškodenia dolného motorického neurónu:
- svalová slabosť (paréza)
- hyporeflexia (znížené reflexy)
- svalová atrofia
- fascikulácie (spontánne, rýchle, nerytmické kontrakcie zväzkov svalových vlákien)
Príznaky poškodenia horného motorického neurónu:
- svalová slabosť (paréza).
- spasticita (zvýšený svalový tonus)
- hyperreflexia (zvýšené reflexy)
- patologické znaky nôh a rúk
Vo väčšine prípadov pre ALS asymetria symptómov.
V atrofovaných alebo dokonca navonok neporušených svaloch, fascikulácie(svalové zášklby), ktoré môžu byť v lokálnej svalovej skupine alebo môžu byť rozšírené.
IN typický prípad nástup ochorenia so stratou hmotnosti svalov thenaru jednej z rúk s rozvojom slabosti addukcie (addukcie) a opozície palca, (zvyčajne asymetricky), čo sťažuje uchopenie palcom a ukazovákov a vedie k narušeniu kontroly jemnej motoriky vo svaloch ruky. Pacient pociťuje ťažkosti pri zdvíhaní malých predmetov, pri zapínaní gombíkov, pri písaní.
Potom, ako choroba postupuje, svaly predlaktia sú zapojené do procesu a ruka nadobúda vzhľad „labky s pazúrmi“. O niekoľko mesiacov neskôr sa vyvinie podobná lézia druhej ruky. Atrofia, postupne sa šíriaca, zachytáva svaly ramena a ramenného pletenca.
V rovnakom čase alebo neskôrčasto dochádza k poškodeniu bulbárnych svalov: fascikulácie a atrofia jazyka, paréza mäkkého podnebia, atrofia svalov hrtana a hltana, ktorá sa prejavuje vo forme dysartrie (poruchy reči), dysfágie (poruchy prehĺtania), slinenie.
Napodobňovať a žuvacie svaly zvyčajne postihnuté neskôr ako iné svalové skupiny. Ako sa choroba vyvíja, je nemožné vystrčiť jazyk, nafúknuť líca a natiahnuť pery do trubice.
Niekedy sa vyvinie slabosť extenzorov hlavy kvôli ktorému pacient nemôže udržať hlavu rovno.
Keď je zapojený do procesu bránice pozoruje sa paradoxné dýchanie (pri inšpirácii žalúdok klesá, pri výdychu vyčnieva).
Nohy zvyčajne najskôr atrofujú predné a bočné svalové skupiny, čo sa prejavuje „visiacou nohou“ a krokovým krokom (pacient zdvihne nohu vysoko a hodí ju dopredu, prudko zníži).
Je charakteristické, že svalová atrofia je selektívna.
- Atrofia sa pozoruje na rukách:
tenara
hypothenar
medzikostné svaly
deltových svalov
- Na nohách sú zapojené svaly, ktoré vykonávajú dorzálnu flexiu chodidla.
- V bulbárnych svaloch sú postihnuté svaly jazyka a mäkkého podnebia.
pyramídový syndróm sa vyvíja spravidla v ranom štádiu ALS a prejavuje sa oživením šľachových reflexov. Následne sa často vyvinie dolná spastická paraparéza. V rukách sa zvýšenie reflexov kombinuje so svalovou atrofiou, t.j. existuje kombinovaná, simultánna lézia centrálnych (pyramídových) dráh a periférneho motorického neurónu, ktorá je charakteristická pre ALS. Povrchové brušné reflexy zmiznú s postupom procesu. Babinský príznak (s čiarkovaným podráždením chodidla, palec na nohe sa ohýba, ostatné prsty sa vejárovito rozchádzajú a ohýbajú) sa pozoruje v polovici prípadov ochorenia.
Môžu sa vyskytnúť zmyslové poruchy. U 10% pacientov sa pozorujú parestézie v distálnych častiach rúk a nôh. Bolesť, niekedy silná, zvyčajne nočná, môže byť spojená so stuhnutosťou kĺbov, dlhotrvajúcou nehybnosťou, kŕčmi v dôsledku vysokej spasticity, s kŕčmi (bolestivé svalové kŕče), depresiou. Strata citlivosti nie je typická.
Okulomotorické poruchy nie sú charakteristické a vyskytujú sa v terminálnych štádiách ochorenia.
Dysfunkcia panvových orgánov nie je typická, ale v pokročilých štádiách sa môže objaviť retencia moču alebo inkontinencia.
Stredná kognitívna porucha(zníženie pamäti a mentálnej výkonnosti) sa prejavujú u polovice pacientov. U 5 % pacientov vzniká frontálny typ, ktorý možno kombinovať s parkinsonizmom.
Charakteristickým znakom ALS je absencia preležanín aj u paralyzovaných pacientov pripútaných na lôžko.
Kdekoľvek sa objavia skoré príznaky ALS, svalová slabosť sa postupne prenáša do väčších častí tela, aj keď pri bulbárnej forme ALS sa pacienti nemusia dožiť úplnej parézy končatín, kvôli zástave dýchania.
V priebehu času pacient stráca schopnosť samostatne sa pohybovať. ALS choroba neovplyvňuje duševný vývoj, najčastejšie však začína hlboká depresia Osoba čaká na smrť. V konečnom štádiu ochorenia sú postihnuté aj svaly, ktoré vykonávajú dýchaciu funkciu a život pacientov je potrebné podporovať umelou ventiláciou pľúc a umelá výživa. Od pozorovania prvých príznakov ALS po smrť trvá 3-5 rokov. Všeobecne sú však známe prípady, kedy sa stav pacientov s jednoznačne rozpoznaným ochorením ALS časom stabilizoval.
KTO MÁ ALS?
Na svete je viac ako 350 000 pacientov s ALS.
ALS je diagnostikovaná u 5-7 ľudí ročne na 100 000 obyvateľov. u viac ako 5 600 Američanov je každoročne diagnostikovaná ALS. To je 15 nových prípadov basy za deň
ALS môže postihnúť kohokoľvek. Miera incidencie (počet nových) ALS - 100 000 ľudí ročne
Menej ako 10 % prípadov ALS je dedičných. ALS môže postihnúť mužov aj ženy ALS postihuje všetky etnické a socioekonomické skupiny
ALS môže postihnúť mladých alebo veľmi starých dospelých, ale najčastejšie je diagnostikovaná v strednej a neskorej dospelosti.
Ľudia s ALS vyžadujú drahé vybavenie, liečbu a neustálu 24/7 starostlivosť
90 % bremena starostlivosti padá na plecia rodinných príslušníkov pacientov s ALS. ALS vedie ku konečnému oslabeniu fyzického, emocionálneho a finančné zdroje V Rusku je viac ako 8 500 pacientov s ALS a v Moskve viac ako 600 pacientov s ALS, hoci toto číslo je oficiálne neustále podhodnotené. Najznámejšími Rusmi s ALS sú Dmitrij Šostakovič, Vladimir Miguľa.
Príčiny ochorenia nie sú známe. Neexistuje žiadny liek na ALS. Došlo k spomaleniu priebehu ochorenia. Predĺženie životnosti je možné pomocou domáceho ventilátora.
Syndrómy, ktoré sú klinicky nerozoznateľné od klasickej ALS, môžu vyplynúť z:
Štrukturálne lézie:
parasagitálne nádory
nádory foramen magnum
spondylóza cervikálny chrbtice
Arnold-Chiariho syndróm
hydromyélia
arteriovenózna anomália miechy
Infekcie:
bakteriálne - tetanus, lymská borelióza
vírusová - poliomyelitída, pásový opar
retrovírusová myelopatia
Intoxikácie, fyzikálne látky:
toxíny - olovo, hliník, iné kovy.
lieky - strychnín, fenytoín
elektrický šok
röntgenových lúčov
Imunologické mechanizmy:
dyskrázia plazmocytov
autoimunitná polyradikuloneuropatia
Paraneoplastické procesy:
parakarcinomatózne
paralymfomatózne
Metabolické poruchy:
hypoglykémia
hyperparatyreóza
tyreotoxikóza
nedostatok folátu,
vitamíny B12, E
malabsorpcia
Dedičné biochemické poruchy:
defekt androgénneho receptora – Kennedyho choroba
nedostatok hexosaminidázy
nedostatok a-glukozidázy - Pompeho choroba
hyperlipidémia
hyperglycinúria
metylkrotonylglycinúria
Všetky tieto stavy môžu spôsobiť symptómy pozorované pri ALS a mali by byť zohľadnené pri diferenciálnej diagnóze.
Na ochorenie neexistuje účinná liečba. Jediný liek, inhibítor uvoľňovania glutamátu riluzol (Rilutek), odďaľuje smrť o 2 až 4 mesiace. Predpisuje sa 50 mg dvakrát denne.
Liečba choroby ALS
Základom liečby je symptomatická liečba:
- Fyzioterapia.
Fyzická aktivita. Pacient by mal v rámci možností zachovať fyzická aktivita Ako choroba postupuje, je potrebný invalidný vozík a ďalšie špeciálne zariadenia.
.Diéta. Dysfágia vytvára nebezpečenstvo vstupu potravy do dýchacieho traktu. Niekedy je potrebné jedlo cez hadičku alebo v gastrostómii.
- Použitie ortopedických pomôcok: krčný golier, rôzne dlahy, pomôcky na uchopenie predmetov.
S kŕčmi (bolestivé svalové kŕče: chinín sulfát 200 mg dvakrát denne alebo fenytoín (Difenin) 200-300 mg / deň alebo karbamazepín (Finlepsin, Tegretol,) 200-400 mg / deň a / alebo vitamín E 400 mg dvakrát denne, rovnako ako prípravky horčíka, verapamil (Isoptin).
.Pri spasticite: baklofén (Baclosan) 10 - 80 mg / deň, alebo tizanidín (Sirdalud) 6 - 24 mg / deň, ako aj klonazepam 1 - 4 mg / deň, alebo memantín 10 - 60 mg / deň.
- Na slinenie atropín 0,25 - 0,75 mg trikrát denne alebo hyoscín (Buscopan) 10 mg trikrát denne.
Ak nie je možné jesť z dôvodu porušenia prehĺtania, aplikuje sa gastrostómia alebo sa zavedie nazogastrická sonda. Včasná perkutánna endoskopická gastrostómia predlžuje život pacientov v priemere o 6 mesiacov.
- o bolestivé syndrómy použiť celý arzenál analgetík. Vrátane narkotických analgetík v záverečných fázach.
- Niekedy určité prechodné zlepšenie prinesú anticholínesterázové lieky (neostigmín metylsulfát – prozerín).
Cerebrolysin vo vysokých dávkach (10-30 ml IV kvapkanie počas 10 dní v opakovaných kúrach). Existuje množstvo malých štúdií preukazujúcich neuroprotektívnu účinnosť cerebrolyzínu pri ALS.
Antidepresíva: Sertalin 50 mg/deň alebo Paxil 20 mg/deň alebo amitriptylín 75-150 mg/deň vedľajšie účinky- spôsobuje sucho v ústach, preto znižuje hypersaliváciu (slinenie), ktoré často trápi pacientov s ALS.
.V prípade porúch dýchania: umelé vetranie pľúc v nemocničnom prostredí sa spravidla nevykonáva, ale niektorí pacienti si kupujú prenosné ventilátory a vykonávajú mechanickú ventiláciu doma.
- Prebieha vývoj na použitie rastového hormónu, neurotrofických faktorov pri ALS.
- V poslednej dobe sa aktívne rozvíja terapia kmeňovými bunkami. Táto metóda sľubuje, že bude sľubná, ale stále je v štádiu vedeckých experimentov.
Lieky, lieky, ktoré pomáhajú spomaľovať ochorenie ALS:
Moderná medicína sa neustále vyvíja. Vedci vytvárajú nové lieky na doteraz nevyliečiteľné choroby. Odborníci však dnes nedokážu ponúknuť adekvátnu liečbu proti všetkým neduhom. Jednou z týchto patológií je ochorenie ALS. Príčiny tohto ochorenia sú stále nepreskúmané a počet pacientov sa každým rokom len zvyšuje. V tomto článku sa bližšie pozrieme na túto patológiu, jej hlavné príznaky a metódy liečby.
všeobecné informácie
Amyotrofická laterálna skleróza (ALS, Charcotova choroba) je závažná patológia nervového systému, pri ktorej dochádza k poškodeniu takzvaných motorických neurónov v mieche, ako aj v mozgovej kôre. Ide o chronické a nevyliečiteľné ochorenie, ktoré postupne vedie k degenerácii celého nervového systému. V posledných štádiách choroby sa človek stáva bezmocným, no zároveň si zachováva duševnú jasnosť a duševné zdravie.
Ochorenie ALS, ktorého príčiny a patogenéza neboli úplne študované, sa nelíši v špecifických metódach diagnostiky a liečby. Vedci ju aj dnes aktívne študujú. Teraz je s istotou známe, že choroba sa vyvíja hlavne u ľudí vo veku 50 až 70 rokov, existujú však prípady skorších lézií.
Klasifikácia
V závislosti od lokalizácie primárnych prejavov ochorenia odborníci rozlišujú tieto formy:
- Lumbosakrálna forma (dochádza k porušeniu motorickej funkcie dolných končatín).
- Bulbárna forma (ovplyvnené sú niektoré jadrá mozgu, čo má za následok charakter).
- Cervikotorakálna forma ( primárne symptómy prejavujú sa zmenami v zvyčajných motorických funkciách Horné končatiny).
Na druhej strane odborníci rozlišujú tri ďalšie typy ochorenia ALS:
Prečo vzniká ALS?
Príčiny túto chorobužiaľ, stále sú zle pochopené. Vedci v súčasnosti identifikujú množstvo faktorov, v prítomnosti ktorých sa pravdepodobnosť ochorenia niekoľkokrát zvyšuje:
ALS choroba. Symptómy
Fotografie pacientov s touto chorobou si môžete prezrieť v špecializovaných referenčných knihách. Všetky majú len jedno spoločné - vonkajšie príznaky ochorenie v neskorších štádiách.
Pokiaľ ide o primárne klinické príznaky patológií, veľmi zriedkavo spôsobujú bdelosť zo strany pacientov. Potenciálni pacienti si ich navyše často vysvetľujú neustálym stresom alebo nedostatkom odpočinku od pracovnej rutiny. Nasledujú príznaky ochorenia, ktoré sa objavujú v počiatočných štádiách:
- svalová slabosť;
- dysartria (ťažkosti s rozprávaním);
- časté svalové kŕče;
- necitlivosť a slabosť končatín;
- mierne svalové zášklby.
Všetky tieto príznaky by mali každého upozorniť a stať sa dôvodom na kontaktovanie špecialistu. V opačnom prípade bude choroba postupovať, čo niekoľkokrát zvyšuje pravdepodobnosť komplikácií.
Priebeh ochorenia
Ako sa ALS vyvíja? Choroba, ktorej symptómy boli uvedené vyššie, spočiatku začína svalová slabosť a necitlivosť končatín. Ak sa patológia vyvinie z nôh, pacienti môžu mať ťažkosti s chôdzou, neustále sa potkýnať.
Ak sa choroba prejavuje porušením horných končatín, nastávajú problémy s vykonávaním najzákladnejších úloh (zapínanie košele, otáčanie kľúča v zámku). 
Ako inak možno rozpoznať ALS? Príčiny ochorenia v 25% prípadov spočívajú v lézii medulla oblongata. Spočiatku sú ťažkosti s rečou a potom s prehĺtaním. To všetko so sebou prináša problémy so žuvaním jedla. Výsledkom je, že človek prestane normálne jesť a schudne. Výsledkom je, že mnohí pacienti spadajú do depresie, keďže ochorenie zvyčajne neovplyvňuje
Niektorí pacienti majú ťažkosti s tvorbou slov a dokonca aj s normálnou koncentráciou. Drobné porušenia tohto druhu sa najčastejšie vysvetľujú zlým dýchaním v noci. Zdravotnícki pracovníci už teraz by mali pacientovi povedať o črtách priebehu ochorenia, možnostiach liečby, aby sa mohol vopred informovane rozhodnúť o svojej budúcnosti.
Väčšina pacientov zomiera respiračné zlyhanie alebo zápal pľúc. Smrť nastáva spravidla do piatich rokov odo dňa potvrdenia choroby.
Diagnostika
Prítomnosť tejto choroby môže potvrdiť iba špecialista. V tejto veci zohráva primárnu úlohu kompetentná interpretácia existujúceho klinický obraz u konkrétneho pacienta. Diferenciálna diagnostika ochorenia ALS je mimoriadne dôležitá.
Aká by mala byť liečba?
Bohužiaľ, dnešná medicína nemôže ponúknuť účinnú terapiu proti tejto chorobe. Ako sa dá ALS vyliečiť? Liečba by mala byť zameraná predovšetkým na spomalenie priebehu patológie. Na tieto účely sa používajú tieto opatrenia:

Liečba kmeňovými bunkami
V mnohých európskych krajinách sú teraz pacienti s ALS liečení vlastnými kmeňovými bunkami, čo tiež spomaľuje progresiu ochorenia. Tento druh terapie je zameraný na zlepšenie primárnych funkcií mozgu. transplantované do oblasti poškodenia, obnovujú neuróny, zlepšujú zásobovanie mozgu kyslíkom a podporujú vznik nových krvných ciev.
Izolácia samotných kmeňových buniek a ich priama transplantácia sa spravidla vykonávajú ambulantne. Po terapii zostáva pacient ešte 2 dni v nemocnici, kde špecialisti sledujú jeho stav. 
Počas samotného postupu sa bunky vstrekujú do cerebrospinálnej tekutiny nie je im umožnené samy sa rozmnožovať pomocou mimo tela a reimplantácia sa vykonáva až po podrobnom očistení.
Je dôležité poznamenať, že tento druh terapie môže výrazne spomaliť ALS ochorenie. Fotografie pacientov 5-6 mesiacov po zákroku toto tvrdenie jednoznačne dokazujú. Nepomôže úplne zbaviť sa choroby. Bohužiaľ, existujú aj prípady, keď liečba nedáva vôbec žiadny účinok.
Záver
Prognóza tohto ochorenia je takmer vždy nepriaznivá. Progresia pohybové poruchy nevyhnutne znamená (2-6 rokov).
V tomto článku sme hovorili o tom, čo predstavuje takú patológiu ako ALS. Ochorenie, ktoré sa nemusí dlho prejavovať príznakmi, sa v súčasnosti nedá úplne vyliečiť. Vedci z celého sveta naďalej aktívne študujú túto chorobu, jej príčiny a rýchlosť vývoja a snažia sa nájsť účinný liek.

